Article Text

Abstract
Background TNR encodes Tenascin-R, an extracellular matrix glycoprotein that is primarily expressed in the central nervous system. Loss of TNR impairs cognition, synaptic plasticity and motor abilities in mice, however its role in human neurodevelopment and cognition is less clear.
Methods and results The authors present the case of a child with intellectual disability and transient choreoathetosis. Array genomic hybridisation revealed a homozygous deletion involving only two genes, including TNR. Sequencing TNR in a cohort of 219 patients with intellectual disability did not identify any potential pathogenic mutations.
Conclusion This is the first report of a complete loss of TNR associated with intellectual disability. This study provides evidence of the important role of TNR in brain development and cognition in humans.
- Tenascin-R
- cognition
- intellectual disability
- microarray
- neurology
- genetics
- neurosciences
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Introduction
A growing body of work indicates that disruption of synaptic plasticity explains a large fraction of individuals with intellectual disability (ID).1 ,2 Tenascin-R (TNR) is an extracellular matrix molecule that has been shown to have an important role in certain forms of synaptic plasticity, and in maintaining a balance between excitatory and inhibitory circuits involved in learning, memory and cognition in mice.3–5 Here, we report the identification of a homozygous deletion encompassing TNR in a patient with global developmental delay, transient choreoathetosis and opisthotonic posturing. This case represents the first report of an individual with a complete loss of TNR, and provides evidence of the important role of TNR in cognition and in the development of the human central nervous system.
Methods
Case report
The patient is a female born to a healthy consanguineous (first cousins) Lebanese couple after normal pregnancy. She has one older sister who is healthy. Family history is otherwise unremarkable (figure 1A). The patient was born at term by an uncomplicated repeat Cesarean section. APGAR scores were 7 and 8 at 1 and 5 min, respectively. Birth weight was 2800 g, length was 54 cm and head circumference was 33 cm (25th, 95th and 25th percentile).
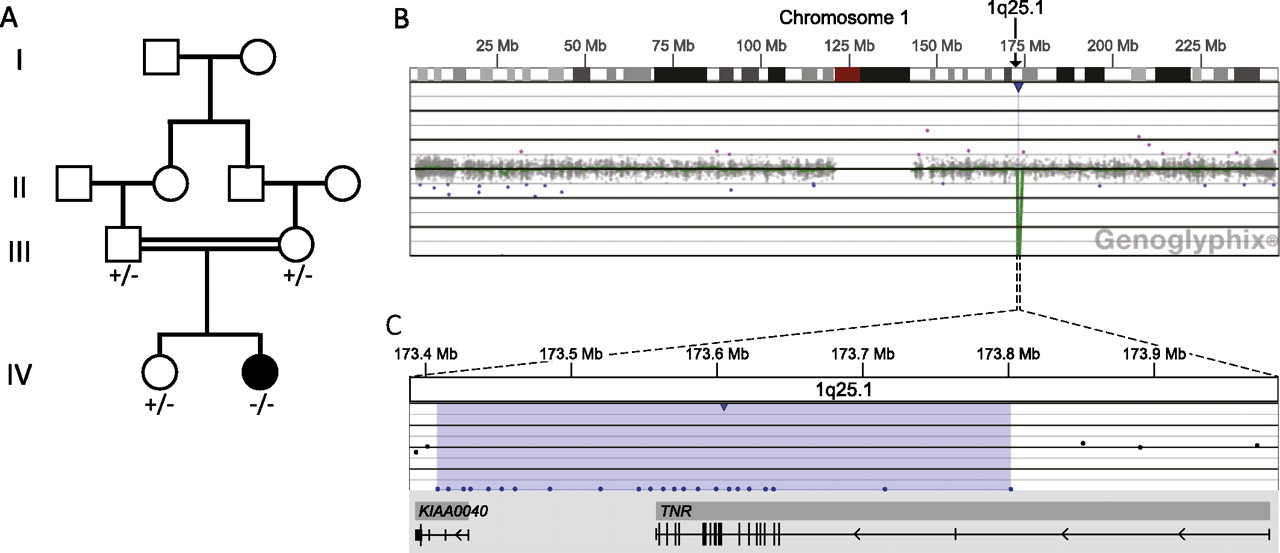
(A) Family pedigree showing segregation of TNR deletion. +, wild-type; −, presence of TNR deletion. (B–C) Microarray analysis shows a two-copy loss of 23 oligonucleotide probes from 1q25.1, approximately 394 kb in size (chr1:173 408 012-173 802 125, UCSC hg18 March 2006 coordinates) resulting in the homozygous deletion of TNR and the non-coding portions of KIAA0040. (B) Probes are ordered on the x-axis and arranged with the most proximal 1q25.1 probes on the left and the most distal 1q25.1 probes on the right. Values on the y-axis represent log2 ratios of patient:control signal intensities. Genes in the region are represented by grey boxes, and vertical black tick marks below the grey boxes show the location of exons. Results are visualised using Genoglyphix (Signature Genomics).
The patient demonstrated opisthotonic posturing several times a day beginning at 4–6 weeks of age. Episodes were more frequent and pronounced during crying and sometimes associated with breath-holding spells. They seldom occurred during sleep, and there were no other abnormal movements or episodes of altered consciousness to suggest seizures. General examination was unremarkable except for a mild synophrys, low-set ears and a right parietal hair whorl. Neurological examination revealed diffuse hyperreflexia. Clonazepam was given, resulting in modest improvement of the opisthotonic posturing. Treatment with levodopa and diphenhydramine did not resolve episodes.
The patient's opisthotonic posturing subsided between 9 and 16 months of age. She then developed choreoathetoid movements in the hands and legs, which resolved by age 4 years. The patient had global developmental delay. She crawled at 30 months, walked at 36 months, used a pincer grasp just before 2 years and started making 2–3-word sentences at 4 years. Examination consistently revealed axial hypotonia, spastic quadriparesis and hyperreflexia. She developed scoliosis, which was treated with nightly use of braces. She acquired microcephaly with a head circumference just below the 2nd percentile.
Head and cervical spine MRI at 3 months of age revealed mild hypoplasia of the inferior cerebellar vermis, but otherwise normal structure and myelination. A comprehensive workup for metabolic disorders including urine purines and pyrimidines, plasma and cerebrospinal fluid amino-acids, urine organic acids and orotic acid, and a white blood cell enzymatic screen for β-galactocerebrosidase, β-galactosidase, β-mannosidase, α-L-fucisidase, β-hexosaminidaseA, arylsulfataseA and glucocerebrosidase was negative. Cerebrospinal fluid neurotransmitter profile and karyotype were unremarkable. Auditory brainstem responses, visual evoked potentials, nerve conduction studies and initial EEG were normal. Electromyography was unremarkable except for signs of chronic muscle disuse. An EEG at 2 years showed spike and wave complexes over the right centrotemporal area, though the patient never developed clinical seizures.
Oligonucleotide microarray and fluorescence in situ hybridisation (FISH)
Oligonucleotide-based microarray comparative genomic hybridisation (aCGH) analysis was performed using a 105K-feature whole-genome microarray (SignatureChip Oligo Solution® version 1.0, custom-designed by Signature Genomic Laboratories, manufactured by Agilent Technologies, Santa Clara, California, USA), according to previously described methods.6 Metaphase FISH analysis was performed using BAC clone RP11-722P23 from the deleted region at 1q25.1 to visualise the abnormality identified in the proband, according to previously described methods.7 Parental and the sister's chromosomes were also studied using metaphase FISH.
ID subjects, TNR DNA sequencing, and variant detection
We recruited 219 subjects with idiopathic ID without growth abnormalities or specific dysmorphic features. From this cohort, only 39 were from consanguineous families that were of diverse ethnic backgrounds. Among the patients from non-consanguineous families, 13 had one other affected sibling, suggesting a possible recessive mode of inheritance. The rest of the patients were sporadic and mostly of French Canadian origin. ID was unexplained in these cases despite standard investigations, including karyotyping, subtelomeric FISH studies or whole-genome aCGH, fragile X testing, and brain CT-scan or MRI. Blood samples were collected from all members of these cohorts as well as from their parents after approval by institutional ethics committees. Genomic DNA was extracted from blood samples.
TNR (Refseq NM_003285.2) coding regions and their intronic flanking sequences were amplified by PCR from genomic DNA, and the resulting products were sequenced. PCR primers targeting the 21 exons of TNR were designed using Exon-Primer from the UCSC Genome Browser (supplementary table S1). Sequencing and variant detection was done as previously described.8
Results
Oligonucleotide microarray and FISH
Oligonucleotide aCGH in the proband revealed a two-copy loss of 23 oligonucleotide probes spanning approximately 394 kb on chromosome 1q25.1 (chr1:173,408,012-173,802,125, UCSC hg18 March 2006 build), and containing only two genes, TNR and KIAA0040: all protein-coding regions of TNR and only the 5′ untranslated region of KIAA0040 were deleted (figure 1B–C). This deletion was confirmed using FISH. Segregation of the deletion was tested by FISH. Both parents and the unaffected sister carried a single-copy deletion of the same region, consistent with the finding that the deletion in the proband was homozygous (figure 2). The homozygous deletion of the protein-coding regions of TNR would result in the lack of a functional TNR protein.

{kind=link}
{kind=link}
FISH visualisation of homozygous TNR deletion. Metaphase FISH in the proband (A), her parents (B–C) and unaffected sister (D) used BAC clone RP11-722P23 from the deleted 1q25.1 region labelled in red. The green control probes are D1Z1 (A–C) and RP11-659D23 (D) from chromosome 1. The absence of a red signal in the proband (A) indicates a homozygous deletion, while the presence of one red signal in the parents (B–C) and unaffected sister (D) indicates they are heterozygous for the deletion (arrows).
Sequencing of a cohort of patients with ID
Sequencing TNR in 219 patients with unexplained ID identified eight missense variations, six of which were found in several patients and reported previously in dbSNP (p.I17V, p.A128S, p.T113I, p.N180H, p.R643K, p.R944P), indicating that they are likely common polymorphisms. The two other heterozygous missenses (p.E201K and G420E113I) were rare and found to be inherited from healthy parents. SIFT and Polyphen predictions indicated that these rare variants, along with the other six common ones, were unlikely to affect protein function. Taken together, these observations suggest that none of these missense changes is likely to be pathogenic. No splicing or truncating mutations were detected in TNR from this screen. (See table S2 for a complete list of TNR variants identified herein).
Discussion
This is the first case description of an individual with a homozygous deletion of the TNR gene. Our patient's phenotype was characterised by global developmental delay, cognitive deficit, transient hyperkinetic movement disorder (opisthotonic posturing and choreoathetosis), central hypotonia and peripheral spasticity. Sequencing TNR in 219 patients with idiopathic ID failed to identify any potential pathogenic mutation, indicating that TNR disruption represents a rare cause of ID. Additional screening of families with ID would be needed to determine the frequency of TNR pathogenic mutations in recessive ID.
Deletions encompassing TNR are extremely rare. Among 38 750 probands tested at Signature Genomics Laboratories, only eight large heterozygous deletions (>5 Mb) that involved TNR along with other genes were detected, and there were no smaller deletions found. The Database of Genomic Variants (http://projects.tcag.ca/variation/) reports three heterozygous deletions encompassing the 3′ end of TNR (3/66741, frequency 4.5×10−5), and there are no exonic deletions in TNR among 2026 control probands in the CNV CHOP database (http://www.cnv.chop.edu). There are no reports of any homozygous deletions of TNR.
Although the deletion in our proband contained in addition to TNR, part of a second gene, KIAA0040, the deletion of TNR is most likely responsible for the phenotype. KIAA0040 encodes a protein whose exact function is currently unknown and whose protein expression was recently detected in colorectal cancer cells.9 Microarray expression experiments suggest that KIAA0040 has little to no expression in the central nervous system making its implication in our patient's pathology unlikely (data from HUGE (http://www.kazusa.or.jp/huge/), Gene Report BioGPS (http://www.biogps.org/), UCSC Genome Browser (http://www.genome.ucsc.edu/)). On the other hand, TNR encodes tenascin-R, an extracellular matrix protein that is expressed mainly in the central nervous system. TNR plays a role in the regulation of neurite outgrowth, neural cell adhesion and cell survival.5 TNR expression is tightly spatiotemporally regulated, especially during the formation of the cortical plate.10 It is produced abundantly by oligodendrocytes and their precursors, and clusters around myelinated axons, particularly around nodes of Ranvier. When myelination is completed, TNR expression decreases with residual expression in a subset of motoneurons and interneurons of the cortex, hippocampus, cerebellum, retina, brainstem and spinal cord. TNR is an important constituent of the perineural nets surrounding inhibitory interneurons.11–13
Homozygous TNR knock-out mice are viable and fertile but show motor and cerebellar abnormalities with deficits in the rota rod testing.13 Behavioural testing reveals severe cognitive deficits, with alteration in associative learning and spatial representation.3 Although gross anatomy and histology are normal, perineuronal nets associated with parvalbumin-expressing GABAergic neurons, which have a role in the regulation of interneuronal excitability, are significantly less developed.13 ,14 In vitro assays of TNR-null hippocampal slices indicate impaired synaptic plasticity with significant defects of long-term potentiation that may underlie the observed impairment in learning and memory.15
The global developmental delay observed in our patient with the homozygous TNR deletion is consistent with a broad dysfunction of learning and memory. Although the opisthotonic posturing and choreaoathetosis observed in our patient have not been described in animal models, it is conceivable that they may be related to deficits in perineural inhibition, especially at the level of the spinal cord and basal ganglia.
In summary, our study reports for the first time a complete loss of TNR in a patient with ID. This observation has implications for our understanding of mechanisms of learning and memory in humans.
Acknowledgments
We would like to thank the patient and her family for their participation, Sylvia Dobrzeniecka for technical assistance and Erin Dodge for assistance in creating the figure. MS holds a clinician-scientist award from the Canadian Institute of Health Research.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data Supplement 1 - Online tables
Footnotes
Competing interests All authors declare no competing interests pertaining to this paper.
Patient consent Obtained.
Ethics approval The ethics approval was provided by the Ethics Committee of the Centre de Recherche de l'Hopital Sainte Justine.
Provenance and peer review Not commissioned; externally peer reviewed.