Article Text

Abstract
Background Mitochondrial DNA (mtDNA) diseases are rare disorders whose prevalence is estimated around 1 in 5000. Patients are usually tested only for deletions and for common mutations of mtDNA which account for 5–40% of cases, depending on the study. However, the prevalence of rare mtDNA mutations is not known.
Methods We analysed the whole mtDNA in a cohort of 743 patients suspected of manifesting a mitochondrial disease, after excluding deletions and common mutations. Both heteroplasmic and homoplasmic variants were identified using two complementary strategies (Surveyor and MitoChip). Multiple correspondence analyses followed by hierarchical ascendant cluster process were used to explore relationships between clinical spectrum, age at onset and localisation of mutations.
Results 7.4% of deleterious mutations and 22.4% of novel putative mutations were identified. Pathogenic heteroplasmic mutations were more frequent than homoplasmic mutations (4.6% vs 2.8%). Patients carrying deleterious mutations showed symptoms before 16 years of age in 67% of cases. Early onset disease (<1 year) was significantly associated with mutations in protein coding genes (mainly in complex I) while late onset disorders (>16 years) were associated with mutations in tRNA genes. MTND5 and MTND6 genes were identified as ‘hotspots’ of mutations, with Leigh syndrome accounting for the large majority of associated phenotypes.
Conclusions Rare mitochondrial DNA mutations probably account for more than 7.4% of patients with respiratory chain deficiency. This study shows that a comprehensive analysis of mtDNA is essential, and should include young children, for an accurate diagnosis that is now accessible with the development of next generation sequencing technology.
- Mitochondrial disease
- Mitochondrial DNA
- Rare mutations
- Patient cohort
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Since the first identification of mitochondrial DNA (mtDNA) defects responsible for human diseases in 1988, it has been increasingly clear that mutations in the mtDNA represent an important cause of neuromuscular disorders.1 ,2 Nevertheless, today, the prevalence of mtDNA disease is still difficult to assess for two main reasons.3 First, large series are extremely rare because of clinical heterogeneity and diagnosis complexity.4 Second, molecular screening is usually restricted to detection of mtDNA deletions and a few common mutations without having accurate data on the prevalence of rare mutations.
Improvements in diagnosis methods led us to study a large cohort of patients from the French Mitochondrial Disease Network, including both children and adults suspected of manifesting a mitochondrial disorder. In our cohort, deletions and common mutations of mtDNA had already been excluded and we performed a comprehensive analysis of the mitochondrial genome in order to answer a number of key questions.
▸ What is the prevalence of rare mtDNA mutations in mitochondrial disorders?
▸ What are the phenotypes associated with these mutations?
▸ Is a comprehensive screening of mtDNA essential for optimal diagnostic approach?
Patients and methods
Study population
Over a 3 year period, 743 individuals suspected of having a mitochondrial disorder were included in the study. All were studied in French referral centres by hospital specialists. A standardised chart including clinical symptoms, imaging and extensive laboratory work was completed for all patients (ie, familial and clinical history, clinical presentation, brain MRI, metabolic screening, mitochondrial enzymes studies, histological and molecular analyses). Age of onset of clinical symptoms ranged from the neonatal period to 74 years of age. The patient population was based on the following inclusion criteria: (1) clinical features suggesting respiratory chain (RC) dysfunction and/or (2) metabolic screening suggesting RC dysfunction and/or (3) isolated or multiple RC complex deficiency and/or (4) histochemical evidence of mitochondrial abnormality in the muscle biopsy and (5) exclusion of mtDNA deletion(s) and m.3243A>G, m.8344A>G and m.8993T>C/G point mutations. Affected individuals presenting with mtDNA depletion or clear mendelian family history were also excluded. Among the 743 patients, 73 satisfied one, 301 two, 275 three and 94 four inclusion criteria. Patients were subdivided into three groups according to their age at presentation: <12 months of age, 1–16 years of age and >16 years of age.
Blood and tissue samples were obtained after parents of affected children and adult patients had given informed consent. Authorisations for individual data protection were obtained.
OXPHOS spectrophotometric measurements
Spectrophotometric studies of oxidative phosphorylation (OXPHOS) complexes and citrate synthase were performed on tissue homogenates and fibroblasts, as previously described.5
Surveyor and MitoChip analysis
The entire human mtDNA was first analysed by the Surveyor method, as previously described.6 ,7 Then, screening was performed with a resequencing chip (MitoChip, GeneChip Mitochondrial Resequencing Array 2.0) according to the manufacturer's instructions (Affymetrix, Inc).8 A custom bioinformatics pipeline was then used for MitoChip analysis, as previously described.9 Sequences were compared with the human mtDNA consensus sequence, Genbank No J01415.2.10
Variants reported in the MITOMAP database (http://www.mitomap.org/MITOMAP) as polymorphisms and present four times or more in the mtDB database (http://www.mtdb.igp.uu.se/) were considered as non-deleterious and excluded from the study.
Statistical analysis
Variables
The variables used refer to clinical data of patients, age at onset (<1 year, 1–16 years, >16 years) and mutation localisation (tRNA genes vs protein coding genes).
Descriptive statistics
Descriptive statistics were used to characterise the population. The unit of analysis was the patient and the analyses were done on the entire population. All variables were categorical.
Multiple correspondence analysis and hierarchical ascendant classification
Analyses were performed on the patient population carrying pathogenic mutations. The unit of analysis was the patient. To explore the genotype–phenotype relationships, we first used multiple correspondence analysis (MCA)11 and next considered the coordinates of the observations on the retained factorial axes as new variables used for hierarchical ascendant classification (HAC).12 MCA allows a graphical representation of the strength of the association between categories of selected variables. The points corresponding to the categories of each variable (eg, age of onset of disease in three categories) are plotted at distances from the origin that are inversely related to the number of patients in a given category. Benzecri's method was used to select the number of axes considered in the analyses.13 MCA was built using age at onset and mutation localisation as supplementary (non-active) variables as they were the variables to explain. The variables with a low frequency (<15%) were excluded from the analysis and Cramer's V test was performed to identify the variables strongly associated with others, which were analysed as non-active variables. To build homogeneous clusters of patients, we performed HAC based on the Ward method. The clustering process was plotted as a dendrogram, with horizontal branches representing the combination of two clusters and vertical branches the degree of dissimilarity between combined clusters. Long distances of the vertical segments indicated large differences between the clusters. Associations between profiles of patients and variables used in the model were tested with Fisher's exact test. Test values were calculated to measure the association between supplementary variables and profiles of patients. We did not use other clustering methods, such as K means or latent class analysis, because of the small size of the population. We chose hierarchical clustering because of the small datasets and the absence of previous assumptions about the number of clusters. All statistical analyses were performed with SAS V.9.3 software (SAS Institute, Cary, North Carolina, USA). A two tailed p≤0.05 was considered statistically significant.
Results
The 743 patients were subdivided into 395 men and 348 women (ratio 1.13 : 1) with an average age of 25 years at the time of the study. Clinical data are shown in the online supplementary table S1 for the entire population, and depending on age of onset. Early clinical symptoms were seen during the first year of life for 356 (48%) patients, between 1 year and 16 years for 192 (25.8%) patients and in adulthood for the remaining 180 (24.2%) patients. In 15 (2%) of 743 cases, age of onset was unknown. Neurological presentations were the most frequent (72.7% of total population). Before 1 year, the most common symptom was psychomotor retardation (55.6%) whereas cerebellar ataxia was commonly seen after 1 year of age or during adulthood (37% and 25%, respectively). Epilepsy and myoclonies were also very common (32.2%) whatever the age of presentation. Muscular involvement was among the second rank of clinical disorders, with chronic progressive external opthalmoplegia (CPEO) mainly seen during childhood and adulthood (27.1% and 32.2%, respectively). Postnatal growth failure was highly represented before 1 year (29.5%). Hypertrophic cardiomyopathy, and liver and kidney deficiencies, were also mainly associated with onset before 1 year (11%, 27.8% and 11.5%, respectively). Optic atrophy was most frequent when the disorder started during childhood (11.5%) whereas peripheral neuropathy was present in 24.4% of patients with adult onset. Paraclinical investigations are presented in the online supplementary table S2. Brain MRI was abnormal in 82.1% of cases before 1 year of age, with a diagnosis of Leigh or Leigh-like syndrome in 43.1% of patients. In patients with adult onset, white matter involvement was the most common pattern (43.8%) whereas posterior fossa atrophy/hypoplasia was the most frequent when onset was during childhood (29.5%). A lactate peak was detected in 60.6% of cases by MRI spectroscopy, mainly in onset before 1 year (68.1%). Hyperlactacidaemia was present in about 50% of patients but hyperlactatorachia was the most common metabolic abnormality (75.3%), mainly during childhood (83.3%) (see online supplementary table S2). Results obtained from tissue biopsies, mainly skeletal tissue, are presented in the online supplementary table S3. The percentage of ragged red fibres (RRF) and COX negative fibres increased with age of onset, reaching 48.9% and 56.9%, respectively, after 16 years of age. In muscle, the biochemical activity of RC complexes was significantly reduced in 59.4% of total biopsies. In liver tissue, a RC deficiency was seen in 53.9% of cases before 1 year of age.
The presence of deletions and common point mutations (m.3243A>G, m.8344A>G and m.8993T>C/G) of mtDNA had previously been ruled out. The first part of the study aimed to search for heteroplasmic mutations by Surveyor analysis. After eliminating the known polymorphisms (list available on request) and, among new sequence variations, the 52 synonymous variants and the 28 nucleotide substitutions in the D loop region (list available on request), a total of 82 different heteroplasmic sequence variations corresponding to putative or known pathogenic mutations were retained (see online supplementary table S4).14–16 The 56 different variants corresponding to putative mutations were not reported in the MITOMAP database and were present less than four times in the mtDB database. Two variants only were found twice. The m.1938A>T variant in MTRNR2 was found in a child who presented with psychomotor delay, epilepsy, ptosis, deafness and white matter lesions on brain MRI. The second patient was an adult presenting with epilepsy, cerebellar ataxia and stroke-like episodes. Neuroimaging revealed patterns of Leigh/MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes) overlap syndrome. In both cases, biochemical analysis revealed no RC deficiency on muscle biopsy. The second variant, m.13604G>C, was in the MTDN5 gene. The first child died at birth and presented a complex I (CI) deficiency in the liver. The second child presented with myopathy, CPEO and deafness. Muscle biopsy showed RRF and COX− fibres but biochemical analysis was not done.
Twenty-six different sequence variations were already known as pathogenic mutations. The characteristics of the 34 corresponding patients (34/743, 4.6%) are presented in table 1. Four mutations (m.9185T>C, m.10191T>C, m.12706T>C and m.13514A>G) were found in two patients, m.14487T>C in three patients and m.13513G>A in four patients. Recurrent mutations were responsible for Leigh syndrome in these patients, except for one, and encoded subunits of CI, except the m.9185T>C variant located in the MTATP6 gene (table 1). This work expands the clinical spectrum of mtDNA disorders, with the example of m.636A>G and m.12236G>A mutations that had been previously reported in non-syndromic hearing loss and that are associated with progressive multisystemic disorders in our cohort.
Characteristics of patients carrying pathogenic mutations identified by Surveyor analysis
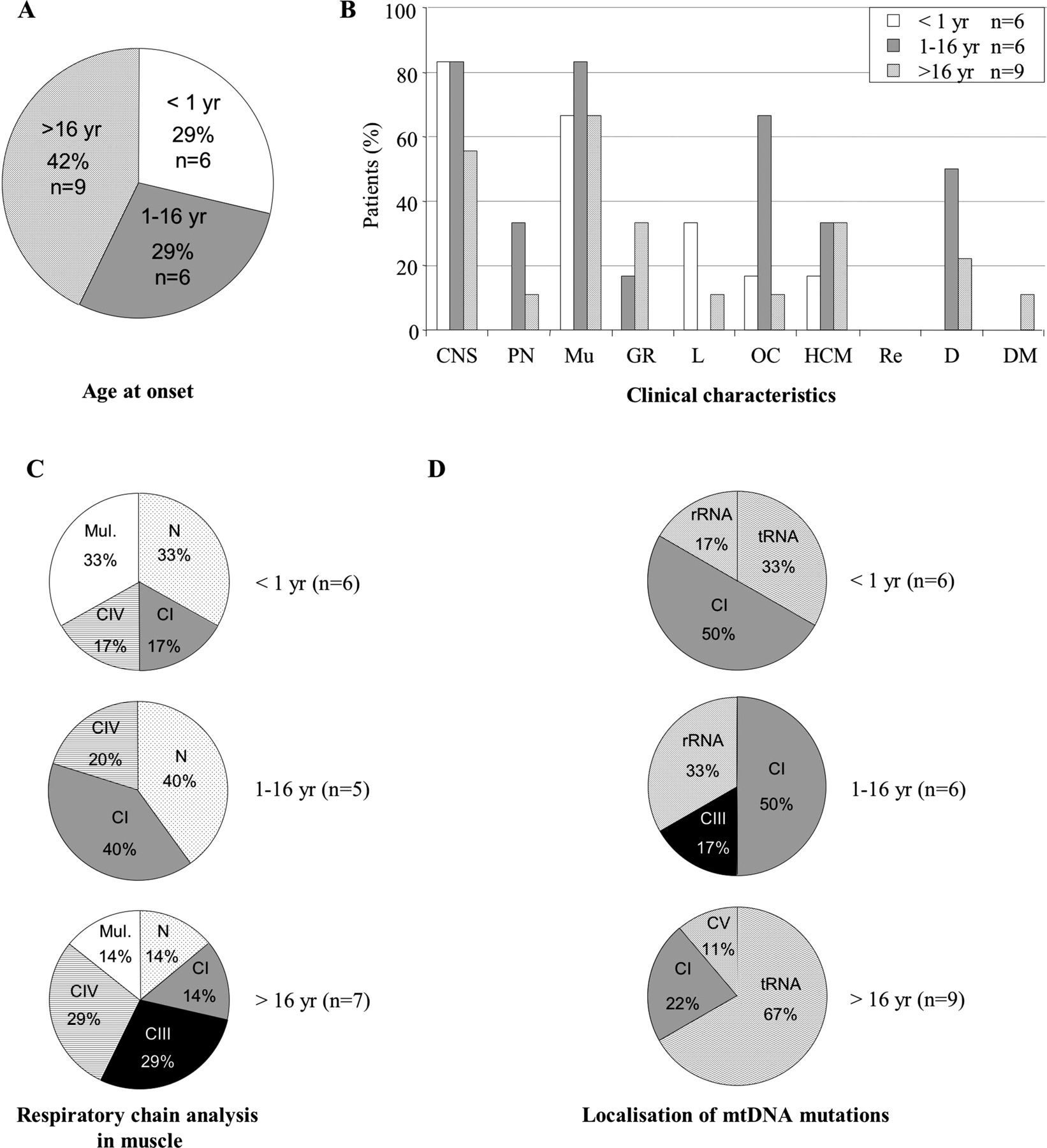
In 74% of cases (25 of 34patients), symptoms started before 16 years of age (figure 1). When onset was before 1 year, all patients presented a multisystemic disorder with abnormal brain MRI, hyperlactataemia and elevation of the lactate to pyruvate ratio. Leigh syndrome was found in 85% of cases (six of seven patients), and usually associated with a CI deficiency in muscle (four of six patients with muscle biopsy, 66.7%). When the onset was between 1 and 16 years of age, ocular symptoms were present in 44.4% of patients (8/18). In adult onset, cerebellar ataxia was found in 44.4% of cases (four of nine patients) and biochemical analysis identified a multiple RC deficiency in muscle in 42.9% (three of seven patients with muscle biopsy). In agreement with the mitochondrial enzyme data, mutations were mainly located in CI genes before 1 year (five of seven patients, 71.4%) and in tRNA genes in adult onset (six of nine patients, 66.7%) (figure 1). Statistical analysis confirmed these results. MCA, used as a step of data reduction, retained 12 active variables: affected maternal relatives, psychomotor retardation, regression, hypotonia, stroke-like episodes, epilepsy, movement disorders, CPEO, optic atrophy, deafness, postnatal growth retardation and Leigh syndrome. The following variables were strongly correlated with others and were added as non-active: cerebellar ataxia, white matter involvement and posterior fossa atrophy or hypoplasia. HAC results showed three distinct profiles (figure 1). The first profile of patients was associated with early onset disease (<1 year) and mutations in protein coding genes. The following variables were overrepresented in this group of patients (p<0.05): psychomotor retardation, hypotonia, optic atrophy, postnatal growth retardation and Leigh syndrome. The second profile of patients was associated with late onset (>16 years) and mutations in tRNA genes. In this profile, affected maternal relatives were common (36%) (p=0.13). We did not find any association for the last profile of patients with a disease onset during childhood (1–16 years). A dendrogram for cluster model is illustrated in the online supplementary figure S1.

Analysis of the population harbouring heteroplasmic pathogenic mitochondrial DNA (mtDNA) mutations according to age at onset. n, number of patients. (A) Age at onset (in years). (B) Clinical characteristics. CNS, central nervous system; D, deafness; DM, diabetes mellitus. GR, growth retardation (intrauterine or postnatal); HCM, hypertrophic cardiomyopathy; L, liver involvement; Mu, muscle; OC, ocular involvement (optic atrophy or pigmentary retinopathy); PN, peripheral neuropathy; Re, renal involvement. (C) Respiratory chain analysis by spectrophotometry. CI, CV, respiratory chain deficiency in complex I, in complex V; mul, multiple respiratory chain deficiency; N, normal. (D) Localisation of mtDNA mutations. CI, CIII, CV, genes encoding subunits of complexes I, III, V; tRNA, genes encoding tRNA. (E) Statistical analyses with hierarchical ascendant classification of pathogenic heteroplasmic mutations found in the 34 patients. The upper right quadrant is complementary to the bottom left quadrant. The numbers on the axes are the scores that represent the contribution of each feature to the overall inertia. Our model (axes 1–3) explains 48% of total inertia. The circles represent the variables. Their size is proportional to the quality of representation of the variables on the plane (axes 1 and 3). Patients with disease onset <1 year are clustered with mutations in protein coding genes, psychomotor retardation, hypotonia, optic atrophy, postnatal growth retardation and Leigh syndrome. Patients with disease onset >16 years are clustered with mutations in tRNA genes and affected maternal relatives.
The second part of the study was to identify homoplasmic mutations using MitoChip analysis in patients without pathogenic heteroplasmic variants. Known polymorphisms and 180 novel variants (140 synonymous and 40 in the D loop) were excluded (list available on request). The online supplementary table S5 shows the 120 different homoplasmic variants found in 131 patients, which correspond to potential and deleterious mutations. Among the 104 variants corresponding to putative pathogenic mutations, four were present more than once (see online supplementary table S5). The m.3228_3229 insA variant in MTRN2 was found in two adults with a mitochondrial myopathy and in one child with Leigh syndrome. The m.8654T>C (p.Ile43Thr) variant in MTATP6 was associated with exercise intolerance and multiple RC deficiency in muscle of an adult patient. The second affected individual was a child with Leigh syndrome. None showed complex V deficiency. The last two variants were in MTCYB. Two adult patients, presenting with exercise intolerance, carried the m.15122A>G (p.Thr126Ala) variant with classical mitochondrial features on muscle biopsy and CIII deficiency in only one of two. The m.15774T>C (p.Val343Ala) variant was associated with two early onset presentations (<1 year). The first child presented with Toni Debré Fanconi syndrome and the second one had a neurological presentation (psychomotor delay, hypotonia and epilepsy). In both cases, RRF fibres were observed on muscle biopsy but we found no RC deficiency.
Sixteen different sequence variations were already known as pathogenic mutations and the characteristics of the 21 corresponding patients (2.8%) are presented in table 2. The m.1555A>G mutation, only seen in a homoplasmic state, was present in three patients with a multisystemic disorder; only one was deaf. Two other mutations were present more than once, and both had been previously found in our study in the heteroplasmic state (m.4317delA, m.13513G>A).
Characteristics of patients with pathogenic mutations identified by MitoChip analysis
Ocular involvement was observed in two-thirds of patients and deafness in half of affected individuals during childhood (figure 2). However, the population size was too small to perform statistical studies.
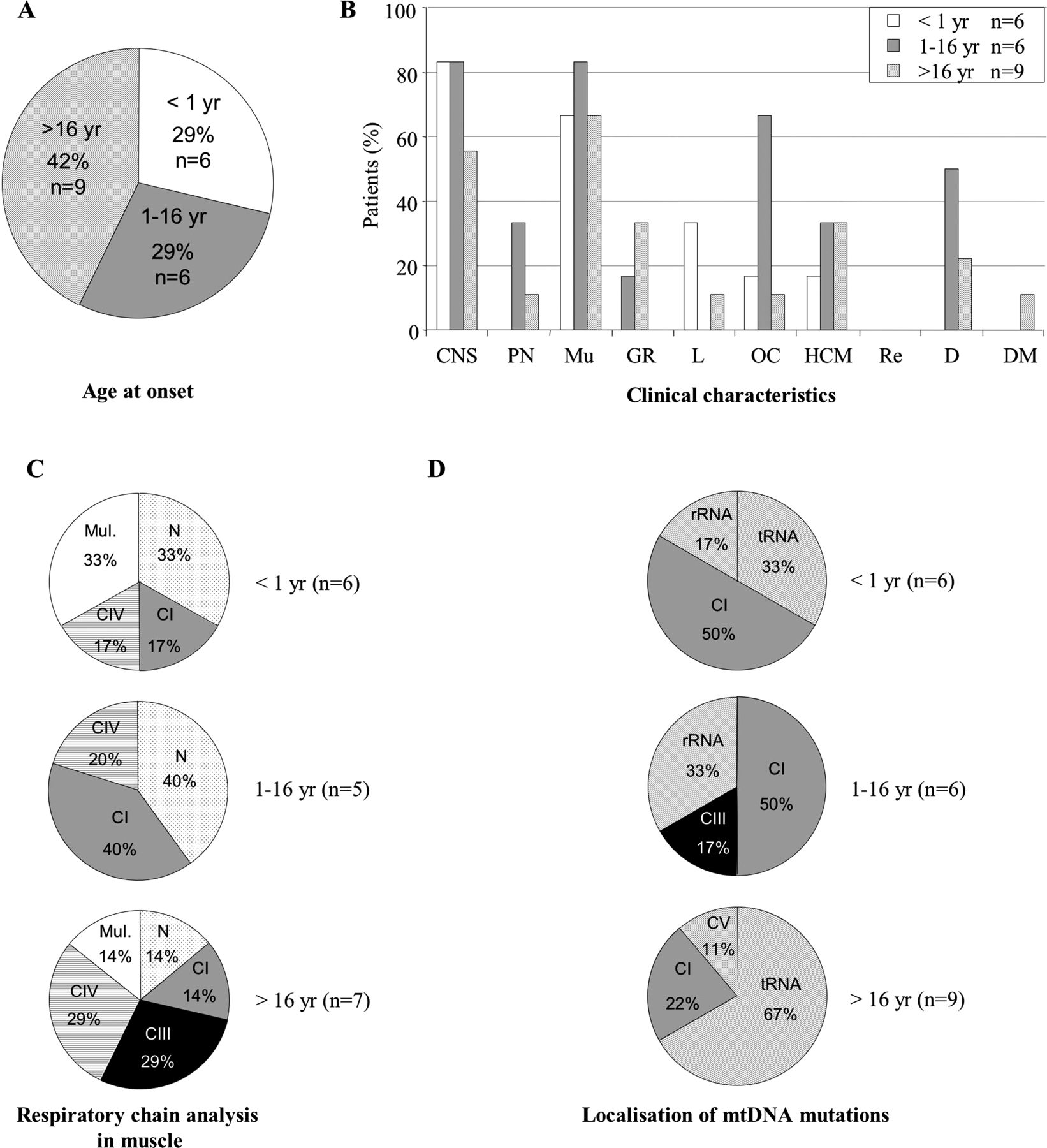
Analysis of the population harbouring homoplasmic pathogenic mitochondrial DNA (mtDNA) mutations according to age at onset. n, number of patients. (A) Age at onset (in years). (B) Clinical characteristics. CNS, central nervous system; D, deafness; DM, diabetes mellitus; GR, growth retardation (intrauterine or postnatal); HCM, hypertrophic cardiomyopathy; L, liver involvement; Mu, muscle; OC, ocular involvement (optic atrophy or pigmentary retinopathy); PN, peripheral neuropathy; Re, renal involvement. (C) Respiratory chain analysis by spectrophotometry. CI, CIII, CIV, respiratory chain deficiency in complex I, complex III, complex IV; mul, multiple respiratory chain deficiency; N, normal. (D) Localisation of mtDNA mutations. CI, CIII, CV, genes encoding subunits of complexes I, III, V; rRNA, genes encoding rRNA; tRNA, genes encoding tRNA.
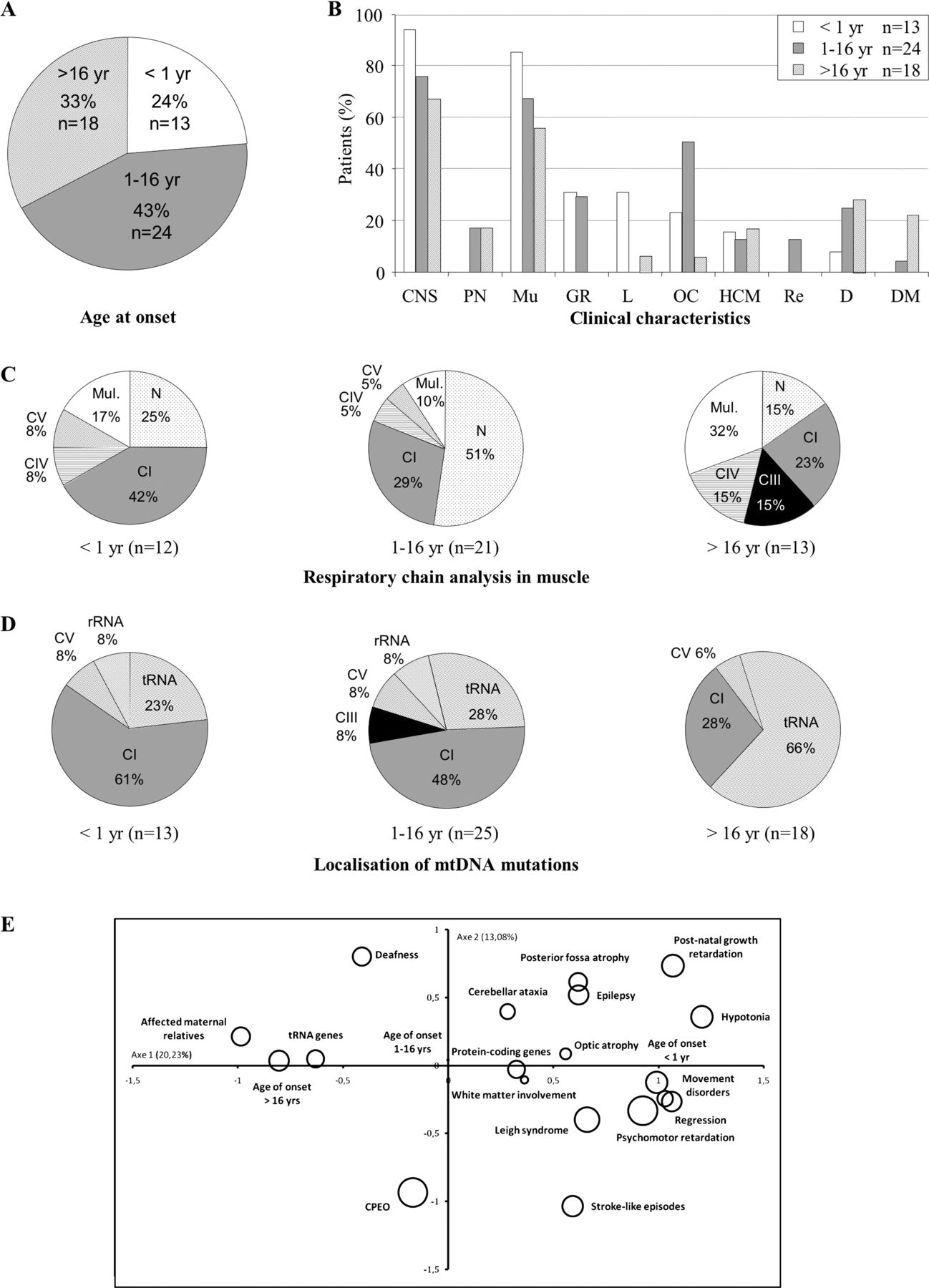
Analysis of all (hetero- and homoplasmic) deleterious mutations confirmed the role of rare mtDNA mutations in paediatric presentations (onset <16 years for 37 of 55 patients, 67.3%) (figure 3). Neuromuscular symptoms were clearly predominant in the 55 patients carrying pathogenic mutations. Liver failure was associated with onset before 1 year, ocular involvement was present in 50% of patients (12/24) in childhood presentations and diabetes mellitus and deafness were found in 20.8% of cases in adult onset disorders (5 of 24 patients). Statistical analysis confirmed previous findings. The same active and non-active variables were retained by MCA. Results of HAC on the 55 patients showed three distinct profiles (figure 3). Early onset disease and mutations in protein coding genes were associated with a profile of patients characterised by psychomotor retardation, regression, stroke-like episodes, movement disorders, Leigh syndrome and white matter involvement. Late onset was associated with mutations in the tRNA genes. Affected maternal relatives were also found predominantly in this second profile (33%), despite being non-significantly associated (p = 0.18). Again, we found no pattern associated with an onset during childhood. A dendrogram for cluster model is illustrated in the online supplementary figure S2.
{kind=link}
{kind=link}
{kind=link}
Analysis of the pooled populations harbouring heteroplasmic or homoplasmic pathogenic mitochondrial DNA (mtDNA) mutations according to age at onset. n, number of patients. (A). Age at onset (in years). (B) Clinical characteristics. CNS, central nervous system; D, deafness; DM, diabetes mellitus; GR, growth retardation (intrauterine or postnatal); HCM, hypertrophic cardiomyopathy; L, liver involvement; Mu, muscle; OC, ocular involvement (optic atrophy or pigmentary retinopathy); PN, peripheral neuropathy; Re, renal involvement. (C) Respiratory chain analysis by spectrophotometry. CI, CIII, CIV, CV, respiratory chain deficiency in complex I, complex III, complex IV, complex V; mul, multiple respiratory chain deficiency; N, normal. (D) Localisation of mtDNA mutations. CI, CIII, CV, genes encoding subunits of complexes I, III, V; rRNA, genes encoding rRNA; tRNA, genes encoding tRNA. (E). Statistical analyses with hierarchical ascendant classification of pathogenic mutations found in the 55 patients. Our model (axes 1–3) explains 45% of total inertia. The circles represent the variables. Their size is proportional to the quality of representation of the variables on the plane (axes 1 and 2). Patients with disease onset <1 year are clustered with protein coding gene mutations, psychomotor retardation, stroke-like episodes, regression, movement disorders, Leigh syndrome and white matter involvement. Patients with late onset are clustered with tRNA gene mutations and affected maternal relatives.
Discussion
In OXPHOS disorders, identification of a causative mutation(s) is critical to confirm the diagnosis and to propose accurate genetic counselling and prenatal diagnosis, but molecular analysis is problematic due to the large genetic heterogeneity of mtDNA related disorders. Primary mtDNA defects include large rearrangements and point mutations. In clinical practice, most suspected patients do not have their entire mitochondrial genome sequenced but are usually tested only for deletions and a small number of common mutations. Several groups have reported that testing for common mtDNA defects in adults with suspected mitochondrial disease can have quite a high diagnostic yield, ranging from 10% to 40% of patients tested.3 ,4 ,17 Testing for common mtDNA mutations has a much lower diagnostic yield in children, ranging from 2–4% to 25%.18 ,19 Recently, looking for 10 mtDNA point mutations in 3168 neonatal cord blood samples revealed that at least 1 in 200 healthy humans harbours a pathogenic mtDNA mutation, although at low mutant heteroplasmy.20 However, to date, the rate of rare mutations is totally unknown. To answer this question, we analysed the whole mitochondrial genome of 743 patients suspected of manifesting a mitochondrial disease after excluding deletions and the most common mutations.
A total of 55 patients (7.4%) harboured pathogenic mutations responsible for an onset disorder before adulthood in 67% of cases and before 1 year of age in 24% of cases. Previous studies have suggested that 50% or more of all patients have adult onset. However, in these studies, the most common mutations (mainly deletions and m.3243A>G) that were mainly found in adult presentations (CPEO, maternally inherited deafness and diabetes (MIDD), etc) were not excluded. The French Mitochondrial Disease Network includes approximately 90% of patients investigated for mitochondrial disease in France. The 743 patients included in the study were doubly selected because they were highly ‘suspect’ of RC disorder, and they did not carry common mtDNA mutations. Our results suggest that in the French population, the clinical presentations that are not associated with common mutations begin mainly before adulthood. Further studies using next generation sequencing technology will be necessary to confirm this point.
Assuming that heteroplasmic and homoplasmic mutations may have different effects, we also analysed both populations separately. As expected, heteroplasmic mutations were more frequent than homoplasmic mutations (4.6% vs 2.8%). Symptoms appeared before the age of 16 years in 75% of patients carrying heteroplasmic pathogenic mutations and in 58% of patients carrying homoplasmic pathogenic mutations. If a number of factors have led to an exaggerated perception that the mtDNA genome is rarely involved in children affected by mitochondrial disorders, our results show that rare mtDNA mutations are a non-negligible cause of paediatric RC deficiency because 6.75% of patients with onset before adulthood (37 of 548) carried rare pathogenic mtDNA mutations.
One of the main results of this study was that the location of mtDNA mutations influenced the age of onset of OXPHOS diseases. We found a significant correlation between mutations in protein coding (mainly CI) genes and early onset disorder (<1 year) on the one hand, and between mutations in tRNA genes and adult onset on the other hand; the hierarchical clustering allowing unambiguous class assignments of patients with early and late onset disease. These results suggest that mutations in tRNA genes are much better tolerated than those that directly affect the functioning of CI whose defect is the most frequently observed. It was previously thought that a mtDNA origin accounted for only 5–10% of paediatric cases.21 However, a recent study of 109 children with isolated CI deficiency identified pathogenic mtDNA mutations in 29% of cases.22 In our study, mutations in MTND genes were found in 11 of 75 patients (14.7%) with an isolated CI deficiency before the age of 16 years but this percentage is likely underestimated because other deleterious mutations were likely present among the 44 variants corresponding to putative mutations that we identified in MTND genes. Among CI genes, MTND5 and MTND6 corresponded to ‘hotspots’ for disease causing mutations, being, respectively, found mutated in 11 (20%) and seven (12.7%) of 55 patients , with Leigh syndrome accounting for the large majority of associated phenotypes.
A total of 167 patients (22.4%) harboured putative pathogenic mutations, with homoplasmic novel variants being more frequent than heteroplasmic variants (14.6% vs 7.8%). Interpretation of the clinical significance of rare variants is challenging due to the highly polymorphic nature of mtDNA. The existing scoring systems to evaluate the pathogenicity of a mtDNA variant consider several parameters, including single fibre PCR or cybrid studies, multiple family reports, segregation within the family, measurable biochemical defect, evolutionary conservation, etc.23 ,24 Collection of data and tools to perform functional studies is in progress but we currently do not have the information needed to precisely define the status of the 160 different variants identified. Nevertheless, 32 patients (4.3%) carried novel variants in protein coding genes with high or medium amino acid conservation and a damaging and not tolerated status according to our in silico analysis using PolyPhen 2 and SIFT, respectively (see online supplementary tables S4 and S5).25 In 15 of 32 patients , the identified variant was correlated with a corresponding biochemical defect (not shown). Regarding tRNA coding genes, mutations were detected in 18 of 22 genes and were found spread all over the structural domains of the corresponding tRNAs. Seventeen of the 36 newly detected variants affect highly conserved nucleotides (Otable 3). Despite the fact that conservation is often poor in picking pathogenicity,28 ,29 this is a criterion that may be taken into account to prioritise future functional studies allowing assessment of tRNA variants using scoring systems.
Novel variants in tRNA genes and tentative prediction of impact according to the level of nucleotide conservation of the affected position
In conclusion, we have reported the first study to determine the prevalence of rare mtDNA mutations in RC disorders. Although further analyses are needed to define precisely the pathogenic or polymorphic nature of all of the novel variants identified, our results show that rare mtDNA mutations account for more 7.4% of patients with mitochondrial disorders. Careful selection of the population studied and the choice of tissues concerned by molecular analysis, which was carried out mainly in muscle (88% of cases), may explain this mutation rate, probably higher if one takes into account some new variants are probably deleterious. In this cohort of patients, we excluded those carrying a deletion or common mtDNA mutations. It is interesting to note that within the French Mitochondrial Disease Network, we have a rate of 8% for these common abnormalities of the mitochondrial genome. The sum of the rates obtained in our network for both common and rare mutations shows how the complete screening of mtDNA is essential. It should be performed for an optimal diagnosis, including in young children, and will now be accessible with the development of next generation sequencing technology that provides a one step approach in detecting common and uncommon point mutations, as well as deletions.
Acknowledgments
We thank all of the patients and referring physicians involved in this study: Professor D Adams, Dr S Attarian, Professor JP Azulay, Dr M Barth, Professor T Billette de Villemeur, Professor D Bonneau, Dr C Boutte, Dr A Cano, Dr S Chabrier, Dr JM Cuisset, Professor P de Lonlay, Professor C Desnuelle, Professor V des Portes, Dr A Echaniz-Laguna, Dr D Feldmann, Dr X Ferrer, Dr M Holder, Dr E Kaphan, Professor D Lacombe, Dr A Lacour, Dr P Laforêt, Dr E Lagrange, Professor P Landrieu, Professor J Lunardi, Dr F Mochel, Professor A Munnich, Dr M-A Nguyen-Morel, Professor J Pouget, Dr M Rio, Dr C Rouzier, Dr C Richelme, Dr S Sacconi, Dr E Salort-Campana, Professor C Verny and Dr A Vershueren. We thank G Augé, S Foustoul, S Gherbi and L Smagghe for technical help.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figure 1
- Data supplement 2 - Online figure 2
- Data supplement 3 - Online table 1
- Data supplement 4 - Online table 2
- Data supplement 5 - Online table 3
- Data supplement 6 - Online table 4
- Data supplement 7 - Online table 5
Footnotes
-
Contributors SB, VP, ASL, CJ, AC, KF, GH, BMdC, SM, CMD, AS, CR, JPB, AR, NA, MG, VD-D, JC, LJ, AD, CE-T, DM, PG, PA-B and BC recruited patients and/or performed biochemical and molecular analyses. HM, NC and ID-Z performed the statistical analysis. VP, XG, HMX, MF and MJF performed the MitoChip data analysis. PR was the leader of the French Mitochondrial Disease Network. KHLQS checked the quality process. SB and CH were responsible for follow-up of the study, synthesis of the data, and produced the tables and figures. CF performed the study of tRNA variants. VP-F was responsible for the study concept and, with SB, obtained funding and wrote the manuscript.
-
Funding This work was made possible by grants to VP-F from the French Ministry of Health (PSTIC 2008, Programme de Soutien aux Techniques Innovantes et Coûteuses).
-
Competing interests None.
-
Ethics approval Authorisation for individual data protection was obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.