Article Text

Abstract
Background Mucopolysaccharidoses (MPS) are monogenic metabolic disorders that significantly affect the skeleton. Eleven enzyme defects in the lysosomal degradation of glycosaminoglycans (GAGs) have been assigned to the known MPS subtypes (I–IX). Arylsulfatase K (ARSK) is a recently characterised lysosomal hydrolase involved in GAG degradation that removes the 2-O-sulfate group from 2-sulfoglucuronate. Knockout of Arsk in mice was consistent with mild storage pathology, but no human phenotype has yet been described.
Methods In this study, we report four affected individuals of two unrelated consanguineous families with homozygous variants c.250C>T, p.(Arg84Cys) and c.560T>A, p.(Leu187Ter) in ARSK, respectively. Functional consequences of the two ARSK variants were assessed by mutation-specific ARSK constructs derived by site-directed mutagenesis, which were ectopically expressed in HT1080 cells. Urinary GAG excretion was analysed by dimethylene blue and electrophoresis, as well as liquid chromatography/mass spectrometry (LC-MS)/MS analysis.
Results The phenotypes of the affected individuals include MPS features, such as short stature, coarse facial features and dysostosis multiplex. Reverse phenotyping in two of the four individuals revealed additional cardiac and ophthalmological abnormalities. Mild elevation of dermatan sulfate was detected in the two subjects investigated by LC-MS/MS. Human HT1080 cells expressing the ARSK-Leu187Ter construct exhibited absent protein levels by western blot, and cells with the ARSK-Arg84Cys construct showed markedly reduced enzyme activity in an ARSK-specific enzymatic assay against 2-O-sulfoglucuronate-containing disaccharides as analysed by C18-reversed-phase chromatography followed by MS.
Conclusion Our work provides a detailed clinical and molecular characterisation of a novel subtype of mucopolysaccharidosis, which we suggest to designate subtype X.
- phenotype
- human genetics
- genetics
- pediatrics
- orthopedics
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Data supporting this paper are provided within the article and Supplementary file. Any additional data not compromised by ethical issues will be made available upon request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Glycosaminoglycans (GAGs) are essential components of the connective tissue. They consist of disaccharide units, each composed of a uronic acid (glucuronate or iduronate) and an N-acetyl-hexosamine (glucosamine or galactosamine), which are further modified by N-sulfation and O-sulfation. Mucopolysaccharidoses (MPS) result from enzymatic defects in the stepwise degradation of sulfated GAGs, leading to lysosomal storage of heparan sulfate (HS), chondroitin sulfate (CS), dermatan sulfate (DS), keratan sulfate (KS), or their degradation intermediates.1 This typically results in multisystem disorders with variable manifestations including skeletal dysplasia (earlier termed ‘dysostosis multiplex’), short trunk disproportionate short stature, coarse facial features, corneal and lens opacity, retinopathy, hypoacusis/hearing loss, valvular heart disease, cardiomyopathy, hepatosplenomegaly and, in some cases also neurodegeneration (OMIM: PS607014). Up to now, eleven different enzymes have been linked to MPS. Classification into different MPS subtypes was made clinically, depending on the phenotype and affected organ systems, followed by the assignment to specific enzyme defects. There is considerable overlap of the skeletal MPS phenotype with other disorders affecting the skeletal system, 461 of which have been summarised in the Nosology and Classification of Genetic Skeletal Disorders in 2019.2 As elevated urinary GAG excretion is a diagnostic hallmark of MPS, total GAG analysis using dimethylmethylene blue (DMB) or uronic acid-based spectrophotometry and quantification of DS, HS and KS by liquid chromatography (LC) are commonly used methods for MPS screening in urine. Leucocyte enzyme activity assays and genetic testing can be used for specification and confirmation of respective MPS subtypes.1 3
Among other enzymes, several sulfatases are involved in the degradation of GAGs.4 The human lysosomal arylsulfatase K (ARSK (OMIM:*610 011)) was identified by computational analysis of the human genome, based on its sulfatase-specific amino acid sequence at the catalytic site (sulfatase signature CXPSR).5 As in all human sulfatases, the conserved cysteine within this sequence undergoes post-translational modification and is converted into an α-formylglycine residue, which is essential for the enzyme activity of all human sulfatases.6 ARSK was found to remove the 2-O-sulfate group from 2-sulfoglucuronate, which led to its other designation ‘glucuronate 2-sulfatase’ (GDS). Glucuronate-2-O-sulfation occurs in HS, DS and CS and, during degradation, is selectively removed by ARSK (online supplemental figure S1).7 As sulfatases show no functional overlap and high substrate specificity,8 ARSK deficiency was discussed as a possible cause of a yet unidentified MPS subtype.9
Supplemental material
A recently published Arsk-deficient mouse model showed a mild lysosomal storage phenotype.10 Glycan reductive isotope labelling-LC/mass spectrometry (MS) revealed HS and CS accumulation in various tissues. Otherwise, the mouse model was characterised by a slight but non-significant elevation of urinary GAG excretion in the absence of skeletal or neurological abnormalities, however, a mild behavioural phenotype was observed.10
Hence, although experimental data suggested an association of biallelic Arsk-variants with an MPS phenotype in mice, no association has yet been established in humans. This is the first report with a comprehensive phenotypical evaluation of four individuals from two unrelated families with ARSK (GDS) deficiency.
Material and methods
Subjects and enrolment
In both families, parents were consanguineous and had two affected children with skeletal dysplasia, resembling spondyloepiphysial dysplasia. The first, female child of family 1 was designated as subject 1 (S1), the second, male child as subject 2 (S2). In family 2, the first two children were unaffected. The third and the fourth male children were indicated as subjects 3 (S3) and 4 (S4), respectively.
Family 1 was referred for genetic analysis from a specialised orthopaedic centre in Austria. Following the identification of ARSK as a candidate gene in family 1, a search for additional affected individuals was carried out using GeneMatcher,11 which identified the second family in India.
Written informed consent for publication was obtained from both families. Explicit permission to publish photographs was obtained for S1 and S2.
Clinical investigations
Medical history and family history were taken during an onsite visit in all 4 subjects (S1–S4).
Reverse phenotyping was performed in subjects 1 and 2 after identifying the ARSK variant to search for further symptoms of MPS, including slit-lamp examination and funduscopy, audiometry, cardiac and abdominal ultrasound, neurological examination, and dermatological assessment. Subjects 3 and 4 have had a thorough assessment of their skeletal phenotype, but no complementary investigations were possible as the patients were not available for follow-up examinations.
Laboratory investigations
A peripheral blood smear was performed with Giemsa staining and was investigated using light microscopy.
GAG excretion in urine (24-hour collection and random sample) was analysed quantitatively with uronic acid and DMB in S1, respectively, and DMB in S2. High-resolution electrophoresis of urinary GAG excretion was performed using standard methods.12 13
Urine and plasma GAGs were measured using a multiplex assay with enzymatic digestion of HS, DS and KS followed by quantification of specific disaccharides by LC-MS/MS as described previously.14
Genetic analysis
In family 1 exome sequencing was performed in S1, using DNA from leucocytes in a diagnostic setting. DNA was extracted using the QIASymphony DSP DNA Midi Kit on a QIASymphony SP instrument (QIAGEN, Hilden, Germany). Nextera DNA Flex Library Prep Kit was used for library preparation and sequencing was performed on a NextSeq 550 (Illumina, San Diego, California, USA). Sequence alignment of raw fastq files to the human reference sequence (GRCh37/hg19 assembly) and variant calling was performed with the DRAGEN Germline Pipeline V.3.2.8 on Illumina BaseSpace (https://basespace.illumina.com/). Variant annotation, filtering and prioritisation using human phenotype ontology (HPO) terms (spondylometaphysial dysplasia HP:0002657 and disproportionate short-trunk short stature HP:0003521) was performed using VarSeq V.2.2 (Golden Helix, Bozeman, Montana, USA, www.goldenhelix.com). MPS-associated genes (GALNS [OMIM:*612 222], GLB1 [OMIM:*611 458], GNS [OMIM:* 607 664], GUSB [OMIM:*611 499], HYAL1 [OMIM:*607 071], IDUA [OMIM:*252 800], ARSB [OMIM:*611 542], HGSNAT [OMIM:*610 453], NAGLU [OMIM:*609 701], SGSH [OMIM:* 605 270], IDS [OMIM:*300 823]) were analysed separately in S1 to exclude a known MPS form. Sanger sequencing was performed for segregation analysis of the ARSK variant in S2 and the patients’ parents.
In family 2 exome sequencing of the probands and two unaffected siblings was performed. Genomic DNA was extracted using the standard phenol-chloroform method. Exome was captured using Illumina’s Nextera Rapid Capture Kit followed by massively parallel sequencing using the NextSeq500 Sequencer (Illumina, San Diego, California, USA) with a targeted average coverage depth of 100×. An inhouse pipeline with integrated burrows-wheeler aligner 1 and genome analysis toolkit V.3.6 best practices2 was used for data analysis. Variants were annotated using ANNOVAR3 and allele frequencies derived from 983 in-house exomes of Indian ethnicity, OMIM phenotypes and HPO terms were integrated in the annotation with the help of inhouse Perl scripts. Region of homozygosity (ROH) analysis was done with the help of FILTUS tool4 using exome data of affected and unaffected siblings. As the family has two affected siblings and reported consanguinity, a pathogenic variant was assumed in the shared homozygous region. The ARSK variant was validated by Sanger sequencing.
Generation of ARSK and ARSG constructs
Different ARSK constructs were generated to prove that the detected variant in family 1, NM_198150.2:c.250C>T, p.(Arg84Cys), results in reduced enzymatic function with a specific enzyme assay.
Wild type-ARSK (ARSK-WT) and ARSK-Cys80Ala constructs were used for comparison of enzymatic function, as normal function was defined by the WT-construct and loss of sulfatase activity had previously been shown for the Cys80Ala-construct.9 An additional construct with the enzyme arylsulfatase G (ARSG[OMIM:* 610 008]), another human sulfatase, was used to show substrate specificity of ARSK.15 All constructs used in this study include a C-terminal histidine-tag (RGSHHHHHH-tag).
The ARSK-Arg84Cys (c.250C>T) and ARSK-Leu187Ter (c.560T>A) constructs were derived by site-directed mutagenesis according to the QuickChange mutagenesis protocol (Agilent Technologies) using the following mutagenesis primers: ARSK_c.250C>T_f:CAATTTGTTGCCCATCATGCGCAGCAATGTG
ARSK_c.250C>T_r:CACATTGCTGCGCATGATGGGCAACAAATTG
ARSK_c.560T>A_f:GACAAAGCAGTAAACTGGTAAAGAAAGGAAGC
ARSK_c.560T>A_r:GCTTCCTTTCTTTACCAGTTTACTGCTTTGTC
The sequences of the resulting constructs were validated by Sanger sequencing.
Transfection and immunoblotting
ARSK-WT, ARSK-Arg84Cys, ARSK-Leu187Ter, ARSK-Cys80Ala and ARSG (N-sulfoglucosamine 3-O-sulfatase) were transiently expressed in HT1080 cells by polyethylenimine (PEI) transfection protocol. Cells were harvested 48 hours after transfection and lysed in phosphate-buffered saline (PBS)/0,5% TX100, sonication was performed on ice (3×10 s), and homogenates were obtained by centrifugation (15000 g, 4°C). Protein determination was performed by detergent compatible (DC) assay (BioRad). Homogenates (50 µg of total protein) were analysed by immunoblotting on polyvinylidene difluoride (PVDF) membrane and antibodies directed against ARSK (biorbyt 160024), ARSG (biorbyt 318995) and GAPDH (Santa Cruz sc-25778, FL-335) as loading control.
Western blot analysis
The effect of the loss-of-function variant, NM_198150.2:c.560T>A, p.(Leu187Ter), in family 2 was investigated by western blot analysis. We used cell lysates (50 µg of total protein) after PEI-mediated transient transfection of HT1080 cells with ARSK-WT, ARSK-Arg84Cys and ARSK-Leu187Ter, respectively.
Glucuronate-2-sulfatase activity assay
The disaccharide 2-sulfoglucuronate-N-acetyl-glucosamine (G2A0) was pre-labelled with the fluorescent dye 2-aminoacridone (AMAC).10 AMAC-labelled G2A0 (12,5 nmol) was incubated with 100 µg protein of the appropriate homogenates in a final volume of 52,5 µl in 250 mM ammonium-acetate buffer pH 4.6 for 24 hours at 37°C. After centrifugation (15000 g, 4°C), the samples were analysed by C18-reversed-phase (RP)-chromatography in ammonium acetate buffer (60 mM, pH 5.6) with a flow rate of 1 mL/min with the Ettan LC system (GE Healthcare). The saccharides were eluted and fractionated with an acetonitrile gradient, in which AMAC-labelled disaccharides were detected by ultraviolet (UV) absorbance at 255 nm. The fractions containing AMAC-mediated fluorescence peaks were vacuum dried, resolved in 25 µl acetonitrile and analysed by nano-electrospray ionization mass spectrometry (ESI-MS) in a negative ion mode.
Results
Biallelic ARSK variants cause a novel MPS phenotype
Genetic testing was performed in S1–S4 between the ages of 14 years and 20 years. Until then, their skeletal dysplasia has not been assigned to a specific genetic disorder. The main radiological abnormalities in all four subjects were thickened calvaria, platyspondyly, anterior inferior beaking of the thoracolumbar vertebrae, broadening of clavicles and ribs, narrowing of lower parts of iliac bones, small epiphyses, metaphysial striae, and hypoplastic carpal bones (online supplemental table S3, figures 1 and 2). Cranial MRI performed due to headache in S1 at 12 years of age showed normal results and absence of enlarged Virchow-Robin spaces.

Phenotype of skull, thorax and spine. Lateral skull radiographs show mildly thickened calvarias of S1 (A, B) and S2 (C–D). S1 and S2 show an open bite (A, C). Radiographs of chest and spine show broad (oar-shaped) ribs (S1 at 17 years (E, F) and S2 at 14 years (H)), broad vertebral bodies, platyspondyly and irregular endplates (S1 at 17 years (G), S3 at 18 years (J)). S1 shows a bell-shaped thorax (E). Radiographs of lateral spine additionally illustrate anterior beaking and irregular vertebral end plates (J). Lateral spine radiograph of the lumbar area shows platyspondyly with scalloping of the posterior end plates (S1 at 12 years (G)). Anterior inferior beaking of the cervical spine of S3 at 20 years (I). S1–S4, subjects 1–4.

Radiological abnormalities of the extremities. Hand radiographs demonstrate short third, fourth and fifth metacarpals, small carpal bones and small epiphyses of lower ends of radius and ulna (S2 at 14 years (A), S4 at 17 years (B), S3 at 18 years (C)). Posterior-Anterior (PA) projection hand radiograph of S2 at 14 years shows defective ossification of the carpal bones associated with retarded bone age. Apparent metaphysial irregularities and dysplasia along the metacarpophalangeal joints (A). PA right pelvis radiograph of S2 at 14 years (D) shows dysplasia and fragmentation of the capital femoral epiphysis associated with acetabular dysplasia. The shaft of the femur is overtubulated. Radiographs of the femora and tibiae demonstrate vertical striae in the metaphyses (S2 at 14 years (E), S4 at 17 years (F)). Tibia and fibula of S2 at 14 years show overtubulation of the shafts (G). Epi-metaphysial dysplasia of the inferior ends of the tibia and fibula. Metaphysial irregularities are associated with metaphysial striation (G). S1–S4, subjects 1–4.
Heart abnormalities in S1 and S2 included cardiac murmur (aortic or mitral), valve disease, thickened leaflets and regurgitation. S1 also showed mild ventricular hypertrophy suggesting mild storage cardiomyopathy, a reduced distensibility of the aorta and an elevated stiffness, as additional known cardiac manifestations of MPS.
All four subjects of this study had normal behaviour and attended a regular school, S1 and S2 at A level. S3 and S4 were not available for follow-up examinations, therefore their behaviour and cognitive function in adulthood remains unknown.
Phenotypical features of S1 and S2 are shown in figure 3 and are summarised in table 1.

Overview of the subjects, their phenotype and health problems. Family 1: (A) pedigree, (C–E) S1 at the age of 16 years, (F, G) S2 at the age of 14 years. Family 2: (B) pedigree. S1–S2 show mild coarse facial features, midface retrusion, full lips (C, F), short-trunk short stature (D, F, G), (relative) macrocephaly (D, F, G), short neck (E, G) and genua valga (F). Hands are shaped normally (H, I). Onset of symptoms in S1 and S2 is depicted in the timeline, age in years (J). MPS, mucopolysaccharidoses; S1–S4, subjects 1–4; WT, wild type. M1=mutation in family 1; M2=mutation in family 2.
Genetic variants and phenotype of patients with ARSK deficiency
Reverse phenotyping in S1 and S2 clearly revealed an MPS phenotype with multisystem involvement. In these two subjects, coarse facial features, short trunk disproportionate short stature and short neck, genua valga, and hip pain were noticed at the end of the first decade. Furthermore, reverse phenotyping revealed mild opacity of the lens and vitreous body. The optical coherence tomography image of the retina of S1 showed loss of interdigitation zone temporal of the macula. Mild aortic valve stenosis and mild left ventricular hypertrophy were noted in cardiac ultrasound in S1 and S2.
Auditory tests revealed normal results in S1 and S2 at the age of 16 years and 14 years, respectively.
Early MPS-related symptoms like macrocephaly at birth (S1 and S2), recurrent ear infections (S2) and sleeping disorder (S2) were initially not attributed to an underlying genetic condition (see timeline figure 3J). Of note, head circumference in S3 and S4 was below the third percentile, proportionate to height at the ages of 18 years and 17 years, respectively.
Absence of vacuolisation in peripheral leucocytes
A peripheral blood smear in S1 and S2 did not reveal abnormal vacuolisation in leucocytes.
Urinary GAG excretion pattern and plasma GAG concentration
Urine analysis in S1 at 11 years of age revealed normal urinary GAG excretion but elevated CS and KS excretion (online supplemental table S1).
These results led to the suspicion of MPS IVA (OMIM:#253000) or MPS IVB (OMIM:#253010) as in these MPS subtypes CS and KS typically show elevated values during childhood.16 The thin-layer chromatography showed a normal pattern of oligosaccharides. Consecutive enzymatic analysis in blood showed normal activity of galactosamine-6-sulfatase and β-galactosidase, thus excluding MPS IVA and MPS IVB.
After identifying the homozygous ARSK variant, quantitative analysis of urinary GAGs with DMB and quantification of DS, HS, KS by high-resolution electrophoresis was repeated in S1 and S2 at the ages of 16 years and 14 years, respectively. The analysis revealed normal results in S1 and borderline total GAG excretion and slightly elevated CS in S2 (online supplemental table S1).
Measurement of urinary GAG excretion by LC-MS/MS analysis in S1 and S2 at the ages of 15 years and 17 years, respectively, showed a threefold and fourfold elevation of specific DS-derived disaccharides in a morning urine sample, respectively (online supplemental table S2), while all other specific GAG-derived disaccharides were in the normal range.
Measurement of plasma GAG concentrations by LC-MS/MS showed no clear difference in the disaccharide pattern of S1 and S2 when compared with 10 control samples, even when analysed in a subgroup of age-matched controls (online supplemental table S2).
Enzyme activity of ß-hexosaminidase, ß-galaktosidase, arylsulfatase A, α-iduronidase, iduronat-2-S sulfatase, sulfamidase, acetylglucosaminidase, GlcNAc-transferase, GlcNAc-6-S-sulfatase, GalNAc-6-S-sulfatase, arylsulfatase B and ß-glucuronidase was measured in fibroblasts of S2 and were all within normal range.
Identification of biallelic ARSK variants in four individuals with a skeletal disorder resembling spondyloepiphysial dysplasia
Exome analysis in S1 revealed a homozygous variant in ARSK, NM_198150.2:c.250C>T, p.(Arg84Cys), located within a 38.4 Mb sized ROH (GRCh37/hg19 Chr5:82940273–121330223). The variant was confirmed in a homozygous state in S2 and a heterozygous state in the patients’ parents (figure 3A).
Targeted variant analysis of known MPS-associated genes in the exome data set of S1 did not reveal pathogenic or likely pathogenic variants in GALNS, GLB1, GNS, GUSB, HYAL1, IDUA, ARSB, HGSNAT, NAGLU, SGSH and IDS.
Exome analysis in family 2 revealed a homozygous nonsense-mutation in ARSK, NM_198150.2:c.560T>A, p.(Leu187Ter), in S3 and S4, located within a ROH of 14 Mb (GRCh37/hg19 Chr5:81721372–95865500). The parents and the unaffected brother were found to be heterozygotes. The unaffected sister did not carry this variant (figure 3B). Both variants have not been reported in variant databases such as ClinVar and Human Gene Mutation Database (HGMD). The detected missense variant c.250C>T, p.(Arg84Cys), results from a C to T substitution, replacing the highly conserved arginine at codon 250 with cysteine, an amino acid with highly different physicochemical properties. The variant affects the sulfatase signature (CXPSR) within the N-terminal sulfatase domain of the protein, which is essential for forming the catalytic site of sulfatases.6 In gnomAD, this variant is listed with a total allele frequency of 0.002129% (6/281810 alleles). The variant c.560T>A, p.(Leu187Ter), is listed with a total allele frequency of 0.0003980% (1/251248 alleles). Of note, no individuals with homozygous loss-of-function variants are represented in gnomAD.
Affected individuals’ ARSK variants cause ARSK deficiency
The arginine at amino acid position 84 in ARSK is part of the highly conserved sulfatase signature 80-CCPSR-84 (online supplemental figure S2) and is crucial for efficient oxidation of the cysteine (Cys80) into a formylglycine residue. This post-translational modification is essential for enzyme activity of all human sulfatases.6
Ectopic expression of the constructs in HT1080 cell lysates revealed comparable expression levels for ARSK-WT, ARSK-Arg84Cys, slightly lower amounts of ARSK-Cys80Ala and lack of protein formation for ARSK-Leu187Ter in western blot analyses, using an ARSK-specific antibody (figure 4D, online supplemental figure S3D). This indicated comparable stability of ARSK-WT and ARSK-Arg84Cys, probable lower stability of ARSK-Cys80Ala, and was compatible with nonsense-mediated mRNA-decay or synthesis of a shortened and thereby highly instable polypeptide as a consequence of the Leu187Ter variant. ARSK-WT-HT1080, ARSK-Arg84Cys-HT1080 and ARSK-Cys80Ala-HT1080 cell lysates only revealed moderate differences in total acid sulfatase activity using the artificial pseudo-substrate p-nitrocatechol sulfate at acidic pH, indicating a high background of the entire set of endogenous lysosomal and non-lysosomal sulfatases (data not shown).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
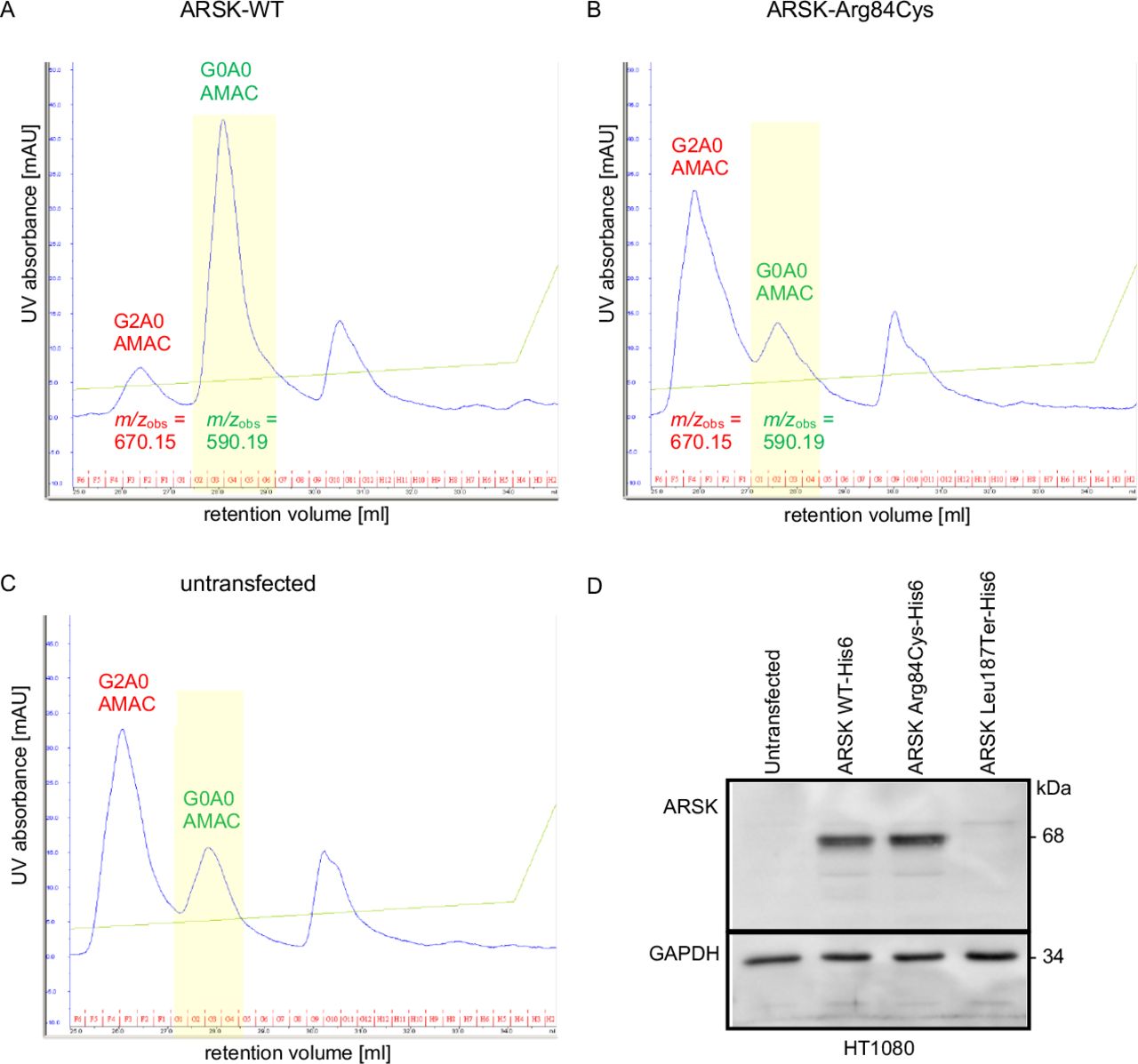
Investigation of the functional consequences of the ARSK variants. ARSK-WT but not ARSK-Arg84Cys desulfates synthetic 2-sulfoglucuronate-N-acetylglucosamine (G2A0) disaccharides (A, B). (A) G2A0 treated with cell lysates expressing ARSK-WT resulted in a minor peak at 26.5 mL representing the 2-O-sulfated educt (m/z 670.15) and a major peak at 28 mL retention volume representing the desulfated product (m/z 590.19), indicating the loss of a sulfate group (highlighted in yellow). Analysis with C18-reversed-phase chromatography. (B, C) Incubation of AMAC-labelled G2A0 disaccharide with ARSK-Arg84Cys cell lysates or with cell lysates of untransfected cells resulted in a main AMAC-peak at 26 mL (m/z 670.15). The G0A0-mediated fluorescence signal remained the minor peak in both samples. Of note, this minor peak in untransfected as well as in ARSK-Arg84Cys transfected cells results most likely from the activity of the endogenous ARSK of the HT1080 cells. The ubiquitous peak in the right of the chromatogram (>30 mL of retention volume) was not analysed in more detail as it was also present in unreacted samples. Western blot analysis (D): Comparable expression levels for ARSK-WT and ARSK-Arg84Cys but lack of ARSK-Leu187Ter in HT1080 cell lysates. This indicates comparable stability of ARSK-WT and ARSK-Arg84Cys and probable nonsense mediated mRNA-decay as a consequence of the Leu187Ter variant. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. WT, wild type.
Therefore, we used the specific ARSK disaccharide substrate G2A0 prelabelled with the fluorescent dye AMAC at the reducing end of the N-acetylglucosamine residue, so that the non-reducing end of the disaccharide was still accessible for enzymatic degradation. After expressing the different ARSK constructs and the ARSG construct, respectively, HT1080 cell lysates were incubated with AMAC-labelled G2A0 and were subsequently analysed by RP HPLC fractionation. Incubation of the AMAC-labelled G2A0 substrate (26 mL retention volume, buffer control) with ARSK-WT homogenates resulted in a main AMAC-mediated detection peak with a retention volume of 28 mL indicating an efficient 2-O-desulfation of the disaccharide leading to the desulfated G0A0 product (figure 4A). However, most other samples (except the buffer sample) including the Cys80Ala variant and the ARSG (N-sulfoglucosamine 3-O-sulfatase) also showed a minor peak at 28 mL (online supplemental figure S3B,C), which was most likely due to endogenous ARSK activity in HT1080 cells rather than residual ARSK activity of ARSK-Arg84Cys and ARSK-Cys80Ala, respectively. MS analysis of indicated peak fractions (figure 4, online supplemental figure S3; m/z values) containing AMAC-labelled disaccharides revealed expected masses of m/z 670.15 for the 2-O-sulfated disaccharide educt peaks and m/z values of 590.19 for the desulfated product within the peaks shifted towards a higher retention volume (figure 4, online supplemental figure S3).
These data clearly demonstrate that the human ARSK-Arg84Cys variant, similar to the previously published ARSK-Cys80Ala construct, is not able to efficiently desulfate the glucuronate-2–0-sulfated disaccharide. Exchange of cysteine and arginine at positions 80 and 84, respectively, leads to strongly reduced sulfatase activity.
Discussion
In this study, we report the identification of biallelic ARSK variants as the underlying cause of a novel subtype of MPS in four individuals of two unrelated families. Affected individuals had skeletal abnormalities and coarse facial features suggestive for MPS, while multisystem involvement with cardiac and ophthalmological findings was only identified by targeted reverse phenotyping.
We demonstrate that the ARSK variant c.250C>T, p.(Arg84Cys) in family 1 leads to ARSK deficiency, proven by a functional GDS assay, whereas the variant c.560T>A, p.(Leu187Ter) in family 2 results in the absence of any ARSK-specific signal in western blot analysis, most probably due to nonsense-mediated mRNA decay.
The effect of ARSK enzymatic deficiency on GAG storage in vivo was recently shown in an Arsk knockout mouse model.10 2-O-sulfated HS was increased in isolated liver lysosomes of Arsk KO mice,10 while no evidence of storage pathology was noted in liver, spleen and kidney of S1–S2 by ultrasound examination. Arsk-deficient mice had a normal skeletal morphology, whereas skeletal abnormalities are the main feature in the human subjects of this study. Although the delineated phenotype in mice is milder and not directly comparable to the human phenotype assessed in this study, the mouse model provides additional evidence that ARSK deficiency leads to a novel subtype of MPS.10
Detailed history and reverse phenotyping of two of the affected individuals (S1 and S2) revealed recurrent bilateral ear infections and sleep disturbance, as noted in 80%–90% of children affected by MPS (type I, II, III, IV).17 Assessment for known ophthalmological features of MPS18 19 revealed mild lens and vitreous opacity and mild pigmentary changes of the retina in S1 and S2. The heart abnormalities which were found in S1 and S2 are in accordance with those typically observed in MPS, like cardiac murmur (aortic or mitral), valve disease, ventricular hypertrophy, thickened leaflets, and regurgitation, reduced distensibility of the aorta and elevated stiffness.20
In S1 total urinary GAG excretion was normal at 11 years of age, which could be due to a decline of urinary GAG excretion with age.21 Electrophoresis in S1 showed elevated excretion of CS and KS, pointing to a possible diagnosis of MPS IV, but enzyme analysis of galactosamine-6-sulfatase (Morquio A) and ß-galactosidase (Morquio B) was normal. Repeated GAG analysis in S1 and S2 at age 16 years and 14 years, respectively, revealed normal results. In addition, the application of the recently developed LC-MS/MS method for the analysis of specific GAG-derived disaccharides revealed a sensitivity for the DMB method of only 90%, underpinning the risk of false-negative results for MPS screening by DMB.14 Urinary GAG analysis by LC-MS/MS in S1 and S2 at 17 years and 15 years of age, respectively, revealed a threefold and fourfold increase of specific DS-derived disaccharides. This is in line with the higher sensitivity of the LC-MS/MS method. The elevated proportion of CS (and KS) detected by electrophoresis in S1 at age 11 years and S2 at age 14 years, respectively, could probably be explained by the overlapping band of accumulated sulfated DS with CS and KS, while no unsulfated DS band was visible on either sample. We speculate that urinary GAG screening by DMB may fail to detect ARSK-associated MPS, which probably contributed to the late identification of this MPS type. Further studies with DMB and electrophoresis, as well as the detection of specific GAG-derived disaccharides by LC-MS/MS, are needed to characterise this novel MPS subtype at the level of accumulating compounds.
In conclusion, the results of this study establish a novel enzyme defect in the group of MPS. ARSK deficiency caused by biallelic loss-of-function mutations or missense mutations affecting the sulfatase signature is associated with childhood-onset disproportionate short-trunk short stature, and skeletal, cardiac and ophthalmological abnormalities. ARSK deficiency should be considered in patients with unclarified skeletal dysplasia or clinically suspected but unassigned MPS, even in the presence of normal urinary GAG excretion. We suggest designating ARSK deficiency as novel subtype X within the group of MPS.
Web resources
GeneMatcher, https://genematcher.org/ gnomAD, http://gnomad.broadinstitute.org/
OMIM, http://www.omim.org
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Data supporting this paper are provided within the article and Supplementary file. Any additional data not compromised by ethical issues will be made available upon request.
Ethics statements
Patient consent for publication
Ethics approval
All studies and investigations were performed according to the declaration of Helsinki principles of medical research involving human subjects. S1 and S2: EK:31–162 ex 18/19; for S3 and S4: IRB of Kasturba Hospital Manipal, India. EIC 201/2010.
Acknowledgments
The authors thank the subjects of this study and their families for participation. The authors also thank Christa Weitzer for performing the abdominal ultrasound and Thomas Weiland for ear, nose and throat examination. The authors thank Christof Pertl for the discussion about the detected tooth and jaw abnormalities, Eduard Paschke for discussion on methods for GAG detection and Tobias Madl and Werner Windischhofer for metabolomic and urine MPS analysis. The authors also thank Henny Rusch for technical assistance and expertise with LC-MS/MS GAGs measurements.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
AAK, KMG and BP are joint last authors.
Contributors Conceptualisation: SV, MRS, TL, KMG, BP. Data curation: SV, JB, GSB, TL, KMG, BP. Formal analysis: SV, JB, JS, FMV, GSB, SGK, TL. Funding acquisition: KMG. Investigation: SV, MI, DA, JS, FMV, LP, DB, HS, AA, KMG, BP. Methodology: GB, MI, DA, JS. Project administration: SV, BP. Resources: MRS, GB, TL, KMG. Supervision: MRS, TL, AA, KMG, BP. Validation: SV, JB, GSB, JS, TL, SGK, BP. Visualisation: SV, LP, TL, KMG. Writing—original draft: SV, BP. Writing—review and editing: SV, JB, MRS, GSB, GB, MI, DA, JS, FMV, SGK, LP, DB, TL, HS, AA, KMG, BP. Guarantor: BP, GK.
Funding Science and Engineering Research Board, Department of Science and Technology, Government of India partially funded this study (work of KMG, Grant Number: SB/SO/HS/005/2014).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.