Article Text

Abstract
T-type channels are low-voltage-activated calcium channels that contribute to a variety of cellular and physiological functions, including neuronal excitability, hormone and neurotransmitter release as well as developmental aspects. Several human conditions including epilepsy, autism spectrum disorders, schizophrenia, motor neuron disorders and aldosteronism have been traced to variations in genes encoding T-type channels. In this short review, we present the genetics of T-type channels with an emphasis on structure-function relationships and associated channelopathies.
- calcium channels
- t-type channels
- cav3 channels
- mutation
- channelopathies
- epilepsy
- autism spectrum disorders
- schizophrenia
- amyotrophic lateral sclerosis
- aldosteronism
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- calcium channels
- t-type channels
- cav3 channels
- mutation
- channelopathies
- epilepsy
- autism spectrum disorders
- schizophrenia
- amyotrophic lateral sclerosis
- aldosteronism
Introduction
Low-voltage-activated Cav3 T-type channels are members of the superfamily of voltage-gated calcium channels.1 T-type channels are widely expressed throughout the nervous, the neuroendocrine and the cardiovascular system2 and are also found in several non-excitable tissues such as osteocytes,3 sperm cells4 and immune cells.5 6 The cellular and physiological processes in which T-type channels are implicated depend primarily on the tissue distribution of the channels. For instance, in the central and peripheral nervous systems, T-type channels play an essential role in shaping neuronal excitability,7–9 whereas they contribute to the release of hormones in the neuroendocrine system.10 11
The essential role of T-type channels in human physiology is emphasised by the existence of channelopathies which are disorders that are caused or enhanced by mutations in genes that encode these channels. Most of T-type channelopathies are transmitted by recessive inheritance or appear sporadic. The clinical manifestations of these disorders depend primarily on dysfunctions of the biophysical characteristics and cell surface trafficking of the channels and can lead to either gain-of-function or loss-of-function.
In this review, we present an overview of human T-type channelopathies and their relationship with the diversity, structure and function of T-type channels. This will be followed by the presentation of the various syndromes for which T-type channels have been linked to, with an emphasis on the structure-function-pathogenicity relationship of mutant Cav3 channels.
Diversity, structure and function of Cav3 channels
Although T-type currents recorded from various native tissues present a common feature illustrated by a low-threshold of activation around −55 mV, they also exhibit several differences in their electrophysiological and pharmacological properties. This heterogeneity is in part explained by the existence of three T-type channel isoforms, Cav3.1,12 Cav3.213 and Cav3.3,14 which in humans are encoded by the genes CACNA1G, CACNA1H and CACNA1I, respectively (figure 1A). This diversity is further enriched by the existence of several channel splice variants. Indeed, alternative splicing of Cav3.1,15–19 Cav3.220–24 and Cav3.325 26 contributes to increase the functional diversity of T-type channels and may also have important pathophysiological implications.

Chromosomal location of human Cav3 channels and their membrane topology. (A) Chromosomal location of human CACNA1G, CACNA1H and CACNA1I genes encoding Cav3.1, Cav3.2 and Cav3.3 channels, respectively. (B) Secondary membrane topology of Cav3 depicting the main structural channel gating determinants.
T-type channels consist on a single Cav3 pore-forming subunit that contains all the structural determinants of channel gating and ion selectivity and permeability (for review see.27 The Cav3 subunit is a relatively large plasma membrane protein of about 260 kDa organised into four hydrophobic domains (DI to DIV), each of them made of six transmembrane helices (S1 to S6) (figure 1B). The voltage-sensing module of the channel is formed by the positively charged arginine/lysine-rich S4 segments,28 while the ion conductivity and selectivity lie on the re-entrant extracellular linkers connecting S5 and S6 segments of each domain, so-called pore-forming loop (P loop).2 The four transmembrane domains are linked together by several intracellular loops connecting the S6 segment of the upstream domain to the S1 segment of the downstream domain, which in combination with the amino and carboxy termini provide hubs for channel regulation by a variety of signalling molecules and other molecular partners including the G-protein βγ-dimer,29 30 CaMKII,31 32 kelch-like 1,33 calcineurin,34 syntaxin-1A,35 stac1,36 CACHD1,37 38 spectrin α/β and ankyrin B39 as well as several ion channels.40 41 In addition, T-type channels undergo several post-translational modifications such as phosphorylation,42 ubiquitination43 and glycosylation,44–49 which contribute to the expression and activity of the channel.
The contribution of T-type channels in particular cellular processes is partly inherent to their unique electrophysiological properties. Voltage-dependent opening of T-type channels occurs at comparatively negative membrane potentials where calcium influx contributes to the depolarisation of the plasma membrane, therefore increasing the opening probability of voltage-gated sodium channels and the propensity of cells to fire action potentials. This aspect is especially relevant in several central neurons including thalamic and hippocampal cells where T-type channels are particularly abundant in dendrites to enhance subthreshold postsynaptic potentials and facilitate the propagation of the electrical signal to the cell body.50 Another way by which T-type channels contribute to neuronal excitability is by forming functional complexes with several types of voltage-activated and calcium-activated potassium channels that allow these channels to operate at subthreshold membrane potentials.51–55 In addition, their fast recovery from inactivation allows T-type channels to generate calcium spikes on brief periods of hyperpolarisation, which leads to the firing of rebound burst of action potentials that support various forms of neuronal rhythmogenesis.56–59 Although a significant fraction of T-type channels is inactivated at most resting membrane potentials of nerve cells, a small fraction remains open to support the passive influx of calcium (termed window current due to the ‘window’ created by the overlap between the activation and inactivation curves of the channels). In nerve cells, this window current has been implicated in the generation of low frequency oscillations observed during sleep patterns60 and is likely to play additional functions especially in non-excitable cells. T-type channels also contribute to several forms of synaptic plasticity.61 Finally, T-type channels are implicated in the low-threshold release of neurotransmitters and hormones, possibly by virtue of their functional coupling with the vesicular release machinery.10 11 Genetic knockout in mice also provided insightful information on the physiological importance of T-type channels. For instance, knockout of Cacna1g has highlighted the role of Cav3.1 in the generation of sinoatrial node pacemaker activity and atrioventricular conduction62 and also their implication in the development of trigeminal neuropathic pain63 and peripheral pain,64 as well as endothelial dysfunction associated with ageing.65 Mice lacking Cacna1h display abnormal coronary function,66 67 decreased susceptibility to cardiac hypertrophy68 and absence seizure,69 decreased peripheral pain signalling70 as well as several neurological symptoms including elevated anxiety and impaired memory.71 72 Mice lacking Cacna1I have provided important information on the implication of Cav3.3 channels in sleep rhythmogenesis.73 74 Finally, several studies from genetic knock out have uncovered a role for T-type channels in the control of myogenic tone.75
Considering that the cellular and physiological functions in which T-type channels are implicated are directly dependent on their electrophysiological properties, it is anticipated that alteration of channel gating caused by mutations will have deleterious consequences. In the next section, we will cover the current state of knowledge of T-type channelopathies and illustrate the links between the structure-function of mutant channels and the pathophysiological features of the associated human syndromes.
CACNA1H (Cav3.2) channelopathies
Idiopathic generalised epilepsy
It is well established that T-type channels play an essential role in the functioning of the thalamocortical circuitry and underlie spike-and-wave discharges that occur during absence seizures.7 69 76–82 This notion is further supported by the observation that thalamic T-type currents are enhanced in several rodent models of absence epilepsy83–85 and genetic overexpression of Cav3.1 channels produces pure epilepsy in rodents.86 Conversely, pharmacological inhibition of T-type currents using pan T-type channel blockers reduces thalamic burst firing and suppresses seizures.87–89 In addition, several pan T-type channel blockers are effective in the treatment of absence seizures in humans90–92 (for recent review see Ref. 81).
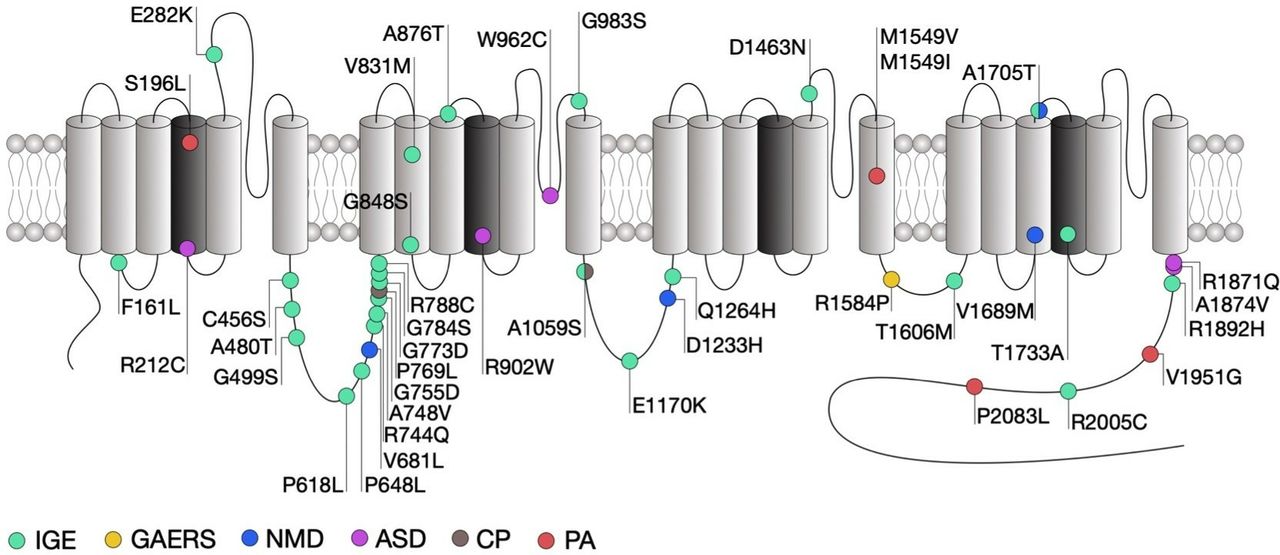
Genetic association studies have identified more than 200 missense variants in the human CACNA1H gene that segregate in patients presenting a range of epilepsy syndromes including childhood absence, juvenile absence, juvenile myoclonic and myoclonic astatic epilepsies as well as febrile seizures and temporal lobe epilepsy that fall under the umbrella term of idiopathic generalised epilepsies (IGE).93–98 It is worth noting that most of these variants have been reported in the Exome Aggregation Consortium (ExAC) suggesting that their contribution to human epilepsies may be rather low or might be dependent on additional genetic and/or environmental factors. Electrophysiological analysis of several of these variants (figure 2) using exogenous expression of mutated Cav3.2 channels in human embryonic kidney cells (HEK293) revealed that these mutations generally produce mild biophysical changes and in some cases do not alter the gating of the channel at all (table 1). This is not completely surprising since the variants examined so far do not concentrate in specific loci that are known to be essential for the gating of the channel, but are rather scattered across the entire channel sequence. However, an interesting study by Zhong and colleagues21 revealed that some of the mutations in Cav3.2 may affect alternative splicing of the channel, which in turn may affect the behaviour of native T-type currents.
Summary of the gating properties of T-type channel variants
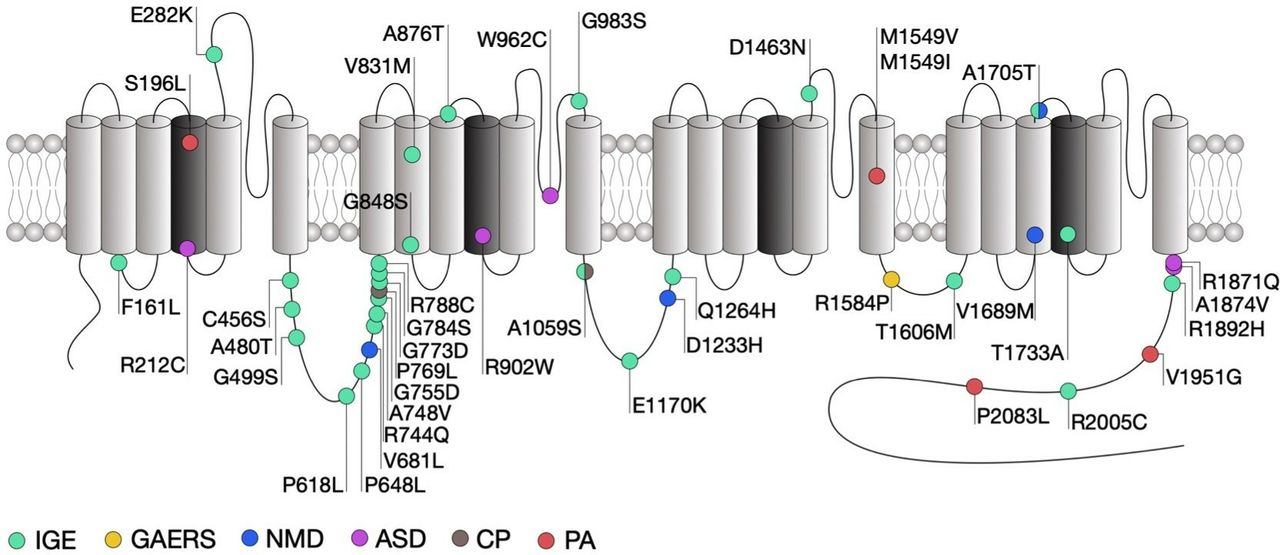
Location of CACNA1H mutations within the secondary structure of Cav3.2 along with their associated syndromes. Only mutations that have been functionally characterised are indicated. Protein reference: UniProt O95180. ASD, autism spectrum disorder; CP, chronic pain; GAERS, genetic absence epilepsy rat from Strasburg; IGE, idiopathic generalised epilepsy; NMD, neuromuscular disorder; PA, primary aldosteronism.
Among the mutations that do affect channel gating, the alterations observed are in general consistent with a gain-of-function of the channel, although in rare exceptions a loss-of-function was observed.99–103 In addition, cell surface expression of the channels may be affected by a subset of mutations within the domain I-II linker region of the channel.104 Intuitively, gain-of-function mutations would increase the propensity of neurons to fire action potentials. This notion is in part supported by computer simulations predicting that several of these mutations would increase neuronal firing and induce neuronal oscillations at similar frequencies as observed during absence seizures.101 In addition, cultured hippocampal neurons expressing the gain-of-function C456S mutation indeed showed increased firing.105 However, studies investigating the influence of Cav3.2 variants in native conditions are too rare to draw a general conclusion on the impact of CACNA1H variants on neuronal excitability.
Despite evident functional effects on Cav3.2 gating which in general are expected to increase neuronal excitability and to potentially drive seizures, only few of the CACNA1H variants identified so far segregate with specific epilepsy phenotypes within families.95 A clear causal mutation linking Cav3.2 to genetic epilepsies was found in the genetic absence epilepsy rat from Strasburg (GAERS) (figure 2).106 This mutation segregates with the occurrence of seizures and causes a gain-of-function of Cav3.2, on the one hand by enhancing the recovery from inaction of the channel23 and on the other hand by enhancing expression of Cav3.2 at the cell surface due to altered association with calnexin.107 Interestingly, biophysical gain of function effects of this mutation selectively manifested themselves in a Cav3.2 splice variant that contained exon 25 which is expressed at high levels in thalamic tissue.23 This may account for the observation that GAERS rats exhibit seizures but do not have other physiological dysfunctions that would be expected from increased T-type channel activity.
Altogether, it remains unclear to which extent CACNA1H variants contribute to human epilepsies. It is likely that these variants only represent a low-risk factor for genetic epilepsies and may only contribute to the disease in combination with other genetic or environmental factors.
Autism spectrum disorder
Autism spectrum disorders (ASD) are neurodevelopmental conditions characterised by impaired social interaction, communication and unusual behaviour. Despite an exceptionally diverse genetic aetiology with hundreds of risk genes identified,108 a subset of high-risk mutations is recurrently found in about 5% of individuals with ASD.109 Several missense mutations in CACNA1H were identified in patients with ASD (figure 2) and were functionally characterised using heterologous expression of mutated Cav3.2 channels.110 Although all these mutations produced several alterations of the channel gating consistent with a loss-of-channel function (table 1), the severity of these alterations appears to be correlated with the location of the mutations in the channel protein. Indeed, and consistent with the observation that the R212C and R920W mutations are located within the voltage sensing region of the channel and neutralise an arginine residue, they produced a depolarising shift of the voltage-dependence of activation of the channel, along with a decreased T-type current. In contrast, the W962C mutation located within the pore-forming loop of the channel did not affect the voltage sensitivity but produced a dramatic decrease of the T-type current, which likely resulted from an alteration of the ionic permeability of the channel. Finally, the R1871Q and A1874V mutations are located in the proximal region of the carboxy terminal region of the channel, a region that is not particularly known to contribute to the gating of the channel and produced only a mild decrease of the T-type current.
As for CACNA1H variants associated with epilepsy syndromes, variants associated with ASD do not segregate with the ASD phenotype, but instead may modify the phenotypic expression. In contrast, several rare de novo gain-of-function mutations with high penetrance were recently identified in CACNA1D and are considered as high-risk factor for ASD and more generally neurodevelopmental conditions.111–114
Neuromuscular disorder
Neuromuscular disorders encompass a wide range of conditions characterised by progressive muscle degeneration and weakness that primarily or secondary impair skeletal muscles and their innervation. For instance, amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a neurodegenerative disorder characterised by the progressive loss of cortical, brain stem and spinal motor neurons, eventually leading to muscle weakness and paralysis. ALS is regarded as a complex genetic disorder with a Mendelian pattern of inheritance in approximately 5%–10% of familial cases,115 but most patients have no discernable family history of the disease which is then referred to being ‘sporadic’ or ‘isolated’ in nature (sALS). However, several genes and loci in apparent sALS cases have been proposed to be associated with an increased risk of the disease or to modify the onset or progression of the disease, which highlights the importance of genetic risk factors.116 Recently, whole exome sequence analysis of case-unaffected-parent trios identified two compound heterozygous recessive missense mutations in CACNA1H (figure 2).117 Functional analysis revealed that these mutations cause a mild alteration of Cav3.2 activity that is consistent with a loss-of-function of the channel (table 1).118 In addition, computer simulations suggested decreased neuronal activity of nerve cells expressing the channel variants.
Although recent studies have reported the expression of T-type channels in motor neurons,119 120 the functional implication of T-type channels in these neurons has yet to be analysed. Increased neuronal excitability has been reported as a hallmark in ALS, where an increase of the sodium conductance and a decrease of axonal potassium currents is observed.121 Considering the role of T-type channels in the control of calcium-activated potassium channels, it is a possibility that decrease of T-type channel activity caused by ALS-associated mutations may contribute to the alteration of potassium currents. In addition, a recent finding demonstrated the role of T-type channels in the maintenance of neuronal progenitor cell viability.122 This aspect will deserve particular attention, especially in the context on neurodegenerative disorders such as ALS. Additionally, a recent study reported the case of a patient with severe infantile onset amyotrophy carrying two inherited heterozygous CACNA1H mutations.123 Functional analysis of Cav3.2 variants were consistent with a loss-of-channel function particularly evidenced by a decreased window current, therefore expending the possible association of CACNA1H with motor neuron diseases.
Chronic pain
It is well established that T-type channels play an essential role in the processing of peripheral nociception and altered expression and altered expression of Cav3.2 has been documented in several chronic pain conditions. For instance, increased activity of Cav3.2 channels in primary afferent fibres is observed in diabetic neuropathy,124 nerve injury,125 irritable bowel syndrome126 and peripheral inflammation43 and is believed to be causally related to the development and maintenance of chronic pain. These gain of function effects in Cav3.2 calcium channels are not linked to mutations in the channel sequence, but instead mediated by altered post-translational modification by deubiqutination43 127–129 and glycosylation processes.45 Recently, two heterozygous missense mutations in the CACNA1H gene were identified in a patient presenting with paediatric chronic pain (figure 2).130 Functional characterisation of these variants using heterologous expression of mutant Cav3.2 channels provided inconclusive results as to the impact of these mutations on the functioning of the channel, mainly due to the observation that the phenotypic manifestations appeared to be dependent on the experimental conditions.
Primary aldosteronism
Primary aldosteronism (PA) is the most common form of secondary hypertension. T-type calcium channels have been implicated in the secretion of aldosterone secretion from the adrenal zona glomerulosa and in situ hybridisation studies combined functional and pharmacological analysis have revealed that Cav3.2 is the main channel isoform generating the T-type current.131 Whole exome sequencing of patients with PA has identified several germline mutations in CACNA1H. Despite an incomplete penetrance, these variants often segregate with the disease132–134 (figure 2). Heterologous expression of the mutated Cav3.2 channels in HEK-293 cells revealed several alterations of the gating of the channel generally consistent with a gain-of-channel function (table 1). In addition, potassium-induced aldosterone secretion is enhanced in several aldosterone-producing adrenocortical cell lines expressing Cav3.2 variants.133 134 This effect may be attributed to a direct potentiation of aldosterone release and/or to an augmented aldosterone production since an increased expression of genes encoding for enzymes involved in the metabolism of aldosterone was also observed in cells expressing Cav3.2 variants. It is important to note that in contrast to CACNA1D mutations that are often associated with severe neurodevelopmental and endocrine disorders,135 136 PA-associated CACNA1H mutations are typically not accompanied with other conditions.
CACNA1G (Cav3.1) channelopathies
CACNA1G has been associated with both cerebellar ataxia and epilepsy. Cerebellar ataxias are clinically heterogenous disorders affecting the cerebellum and cerebellar pathways resulting in impaired coordination. While non-genetic ataxias are caused by acquired conditions or sporadic neurodegenerative disorders, several genes have been associated with hereditary cerebellar ataxias where ion channels are largely represented.137 Among these genes, CACNA1G encoding for the Cav3.1 T-type channel has emerged as a potential contributor and whole exosome sequencing identified a common R1715H variant that segregates in several families with autosomal dominant cerebellar ataxia.138–141 This mutation is located in the IVS4 voltage sensing region of Cav3.1 (figure 3). Consistent with the notion that positively charged residues within the voltage sensing region of the channel are essential for the gating, electrophysiological analysis of the mutant Cav3.1 channel in HEK-293 cells revealed a variety of alterations consistent with a loss-of-channel function (table 1), which is corroborated by computer simulations in deep cerebellar nuclei neurons that suggest a decreased neuronal excitability. Importantly, altered T-type currents were also confirmed in iPSC-derived Purkinje cells from patient carrying the R1715H variant.139 Gain-of-function mutations in CACNA1G have also been identified in patients with childhood-onset cerebellar atrophy.142 Three subjects showed an A961T variant, and one patient had an M1531V substitution. Both of these mutations induced gains-of-function by impairing the inactivation of the channel. A recent case report has implicated CACNA1G mutations in spinocerebellar ataxia type 42. An M1574L mutation was found in three patients from a Chinese family. In addition to ataxia, the clinical phenotype included cerebellar atrophy and brainstem defects.143

Location of CACNA1G mutations within the secondary structure of Cav3.1 along with their associated syndromes. Only mutations that have been functionally characterised are indicated. Protein reference: UniProt O43497. CA, cerebellar ataxia; IDCN, deep cerebellar nuclei; GE, idiopathic generalised epilepsy; TRN, thalamic reticular nucleus.
Mutations in Cav3.1 have been associated with the development of epilepsy. In a cohort of 123 patients with IGEs, 13 CACNA1G variants were identified, including five that led to amino acid substitutions.144 In this study, an A570V substitution was found in a sporadic case of juvenile myoclonic epilepsy, as was an A1089S substitution that was detected in three family members. A biophysical analysis of the biophysical consequences of these mutations did not identify statistically significant effects. Interestingly, it has recently been reported that the CACNA1G gene can act as a modifier of Dravet syndrome induced by defects in the sodium channel Nav1.2,145 146 suggesting that previously identified CACNA1G mutations may also interact phenotypically with other genes, rather than being pathogenic per se.
CACNA1I (Cav3.3) channelopathies
As for CACNA1G, the channelopathies associated with CACNA1I are yet limited and only recently two de novo missense variations were identified in individual with schizophrenia,147 a psychiatric disorder for which the genetic includes a variety of common and rare variants.148 Both mutations are located in the pore-forming region of Cav3.3 (figure 4). While these two variants had no biophysical effect, the A1346H mutation caused a significant decrease of the expression of the channel, possibly by altering glycosylation of Cav3.3.149 In contrast, the T797M variant had no impact on the channel (table 1). Computer simulations in thalamic reticular nucleus neurons also support a decreased neuronal excitability caused by schizophrenia-associated R1346H mutation. In contrast with Cav3.1 and Cav3.2, mutations in Cav3.3 have so far not been associated with epilepsy.150
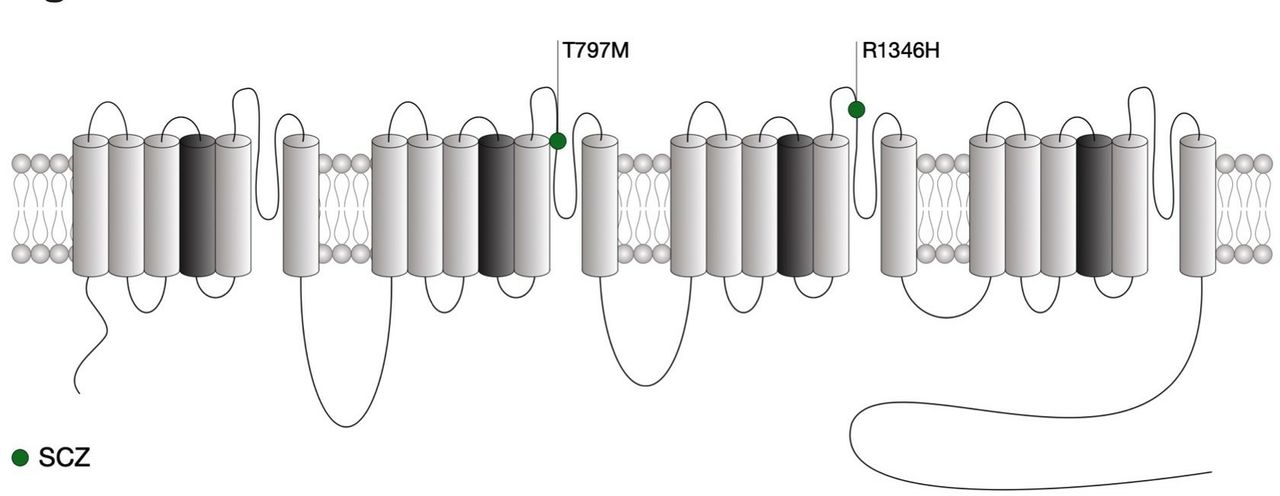
Location of CACNA1I mutations within the secondary structure of Cav3.3 along with their associated syndromes. Only mutations that have been functionally characterised are indicated. Protein reference: UniProt Q9P0×4. SCZ, schizophrenia.
Functional prediction of Cav3 mutations
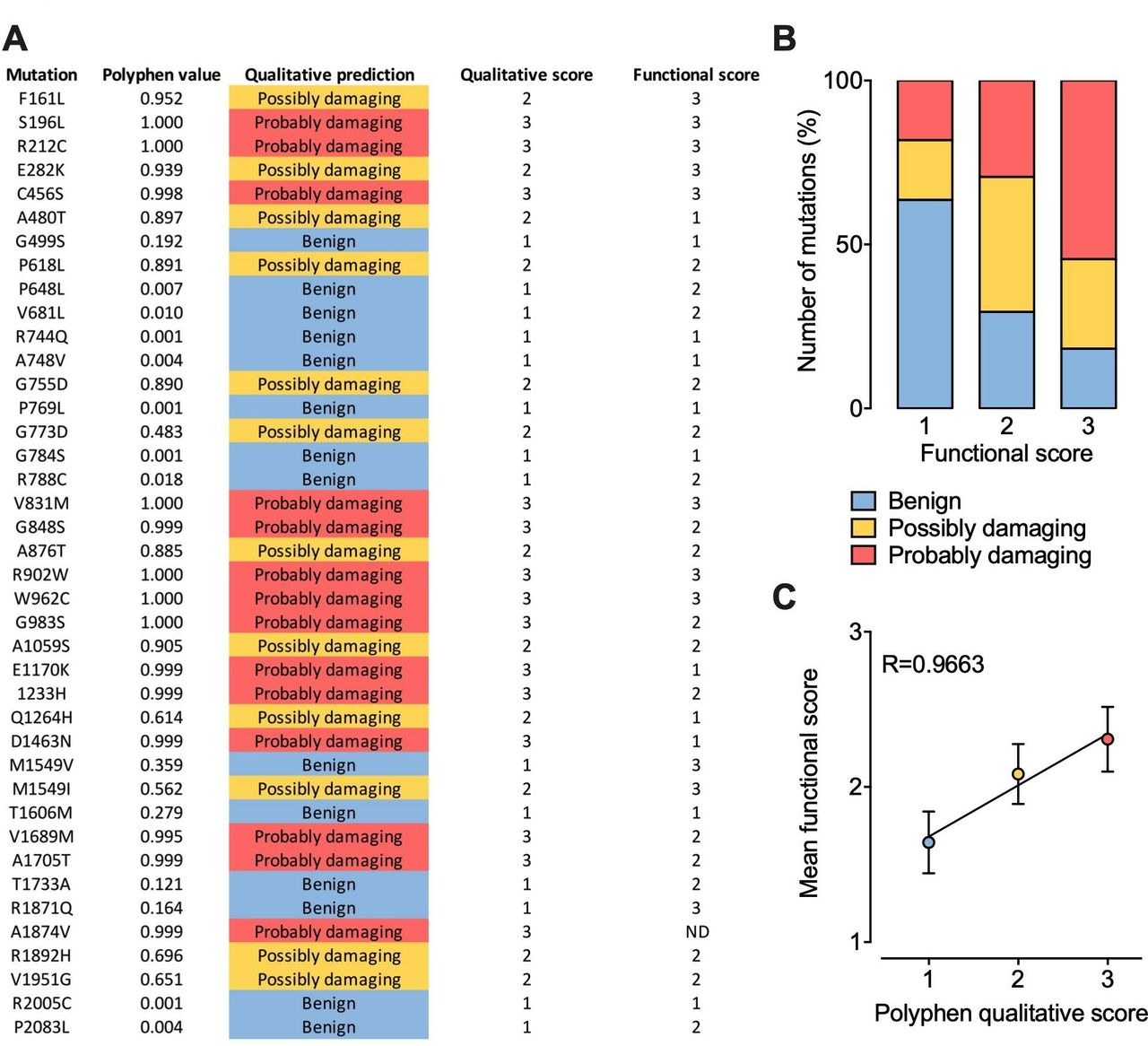
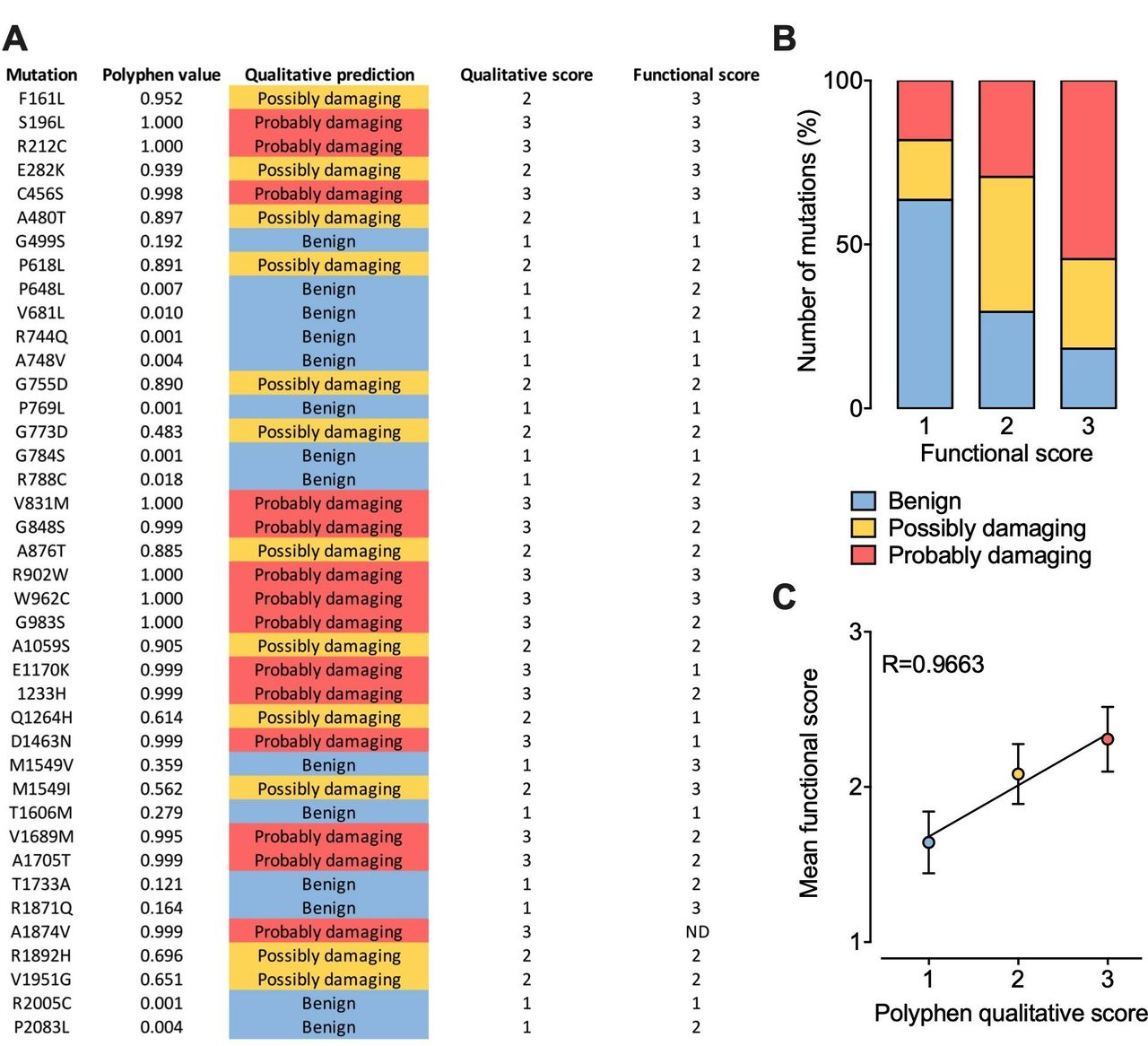
Although the effect of several Cav3 mutations on the functioning of the channel has already been characterised, hundreds of additional variants have yet to be analysed. The question then arises as to whether we could predict their functional impact. Therefore, we collected all functional data currently available for Cav3.2 in order to attribute a functional score for each mutation. The functional score was established as follow: score 1 for mutations producing less than a 2 mV alteration of the voltage-dependence of activation or inactivation or less than a 20% alteration of the kinetics (activation, inaction, recovery from inactivation) or current density compared with the wild type channel; score 2 for mutations producing a between 2 mV and 5 mV alteration of the voltage-dependence or a between 20% and 50% alteration of the kinetics or current density; score 3 for mutations producing more than a 5 mV alteration of the voltage-dependence or more than a 50% alteration of the kinetics or current density. Because functional analyses are often performed using different experimental conditions such as the concentration and nature of the permeating cation which directly affect the gating properties of the channel, the functional score attributed for each mutation is based on the relative biophysical effect of the mutation compared with the gating properties of the wild type channel recorded in the same experimental settings. In parallel, each mutation was probed using PolyPhen-2 algorithm151 that predicts the possible impact of an amino acid substitution on the structure and function of a human protein, providing a qualitative score 1 for benign, 2 for possibly damaging and 3 for probably damaging) (figure 5A). Qualitatively, we observed that higher PolyPhen scores tend to associated with higher functional scores, although a small fraction of mutations predicted to be damaging are not associated with functional alteration and vice versa (figure 5B). When plotting the average functional scores against Polyphen predicted scores, we observed a very strong correlation (R=0.9663), suggesting that in average it may be possible to predict to which extent a mutation might alter the functioning of the channel with a relative degree of certainty (figure 5C).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Prediction of CACNA1H variants on Cav3.2 channel gating. (A) Summary of PolyPhen-2 qualitative prediction and functional experimental score for each Cav3.2 mutations characterised experimentally. (B) Chart representing the proportion of Cav3.2 variants according to Polyphen-2 prediction as a function of their functional score. (C) Plot of the mean functional score as a function of Polyphen-2 prediction.
Conclusion and perspectives
A number of genetic association studies have identified variations in the genes encoding different T-type channels and associated with several neuronal, neuroendocrine and psychiatric syndromes. However, because of the absence of traditional segregation patterns in families in part due to reduced penetrance in adults with the absence of large multiplex families, de novo mutations and/or variable expressivity, many Mendelian traits are likely overlooked. In addition, functional analysis of Cav3 mutants indicate that these mutations generally produce mild alterations of the channel activity, which may be interpreted as a weak evidence of the implication of the channel in the disease. However, several considerations need to be made. First, functional studies are largely predominated by the use of heterologous expression systems that may not entirely reflect the extent to which the mutations affect the functioning of the channel. For instance, although the cellular and physiological aspects controlled by T-type channels are largely dependent on their intrinsic gating properties, recent studies have shown that T-type channels are far more complex than anticipated in terms of their regulation and association with other signalling molecules. The impact of the mutations on these aspects is likely to be overlooked in heterologous expression systems. Second, several Cav3 splice variants have been documented. Therefore, it is worth considering the possibility that the functional expression of the mutations may vary depending on the channel splice variant in which it is reintroduced. For instance, as noted earlier, this notion is supported by the observation that the biophysical expression of the GAERS mutation depends on the Cav3.2 splice variant used.23 Third, it is currently complicated to fully apprehend the long-term impact of small alterations of the channel gating on neurodevelopmental aspects which may have an important impact on the pathogenesis of the disease. Indeed, in addition to their role in neuronal excitability, T-type channels also contribute to several developmental aspects. Finally, the observation that T-type channel mutations are not confined to a particular structural determinant but are rather scattered across the entire protein highlight the need for additional structure-function relationship studies. It is also important to note that mutations in a given gene and producing virtually identical biophysical alterations can lead to different disorders typically without overlap between the diseases. This is, for instance, the case for CACNA1H mutations where a gain-of-function phonotype leads to PA without conferring other disease risk, while similar gain-of-function mutations are associated with increased risk for IGE without concomitant endocrine disorders. This phenotypic heterogeneity suggests that several additional factors such as modifier genes, environmental aspects, allelic variations or complex genetic and environmental interactions are likely to contribute to penetrance and expressivity of these mutations.152
In addition to disorders for which T-type channels have already been implicated, it is likely that other disorders could be caused or influenced by mutations or polymorphisms in T-type channel genes. Indeed, besides being expressed in electrically excitable tissues, T-type channels are also present in several non-excitable cells. For instance, Cav3.1 channels are functionally expressed in immune T cells where they shape the immune response.5 6 T-type channels are also expressed in sperm cells where they participate to the fertilisation of the egg.4 Similarly, T-type channels contribute to calcium signalling in osteocytes.3 This suggests that additional human T-type channelopathies might exist. An important step forward in our understanding of T-type channelopathies will be the identification of modifier genes that are likely to play an important role in the phenotypic expressivity of T-type channel variants.
References
Footnotes
Contributors NW and GWZ analysed the literature and wrote the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.