Article Text

Abstract
Background Distal hereditary motor neuronopathies (dHMN) are a group of genetic disorders characterised by motor neuron degeneration leading to muscle weakness that are caused by mutations in various genes. HMNJ is a distinct form of the disease that has been identified in patients from the Jerash region of Jordan. Our aim was to identify and characterise the genetic cause of HMNJ.
Methods We used whole exome and Sanger sequencing to identify a novel genetic variant associated with the disease and then carried out immunoblot, immunofluorescence and apoptosis assays to extract functional data and clarify the effect of this novel SIGMAR1 mutation. Physical and neurological examinations were performed on selected patients and unaffected individuals in order to re-evaluate clinical status of patients 20 years after the initial description of HMNJ as well as to evaluate new and previously undescribed patients with HMNJ.
Results A homozygous missense mutation (c.500A>T, N167I) in exon 4 of the SIGMAR1 gene was identified, cosegregating with HMNJ in the 27 patients from 7 previously described consanguineous families and 3 newly ascertained patients. The mutant SIGMAR1 exhibits reduced expression, altered subcellular distribution and elevates cell death when expressed.
Conclusion In conclusion, the homozygous SIGMAR1 c.500A>T mutation causes dHMN of the Jerash type, possibly due to a significant drop of protein levels. This finding is in agreement with other SIGMAR1 mutations that have been associated with autosomal recessive dHMN with pyramidal signs; thus, our findings further support that SIGMAR1 be added to the dHMN genes diagnostic panel.
- SIGMAR1 mutation
- distal hereditary motor neuronopathy
- Jerash dHMN
- HMNJ
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Distal hereditary motor neuronopathies (dHMN) are a heterogeneous group of motor neuron disorders characterised by weakness and wasting of the distal muscles of both limbs.1 So far, 30 genes have been associated with dHMN etiopathogenesis that encode proteins involved in a variety of cellular processes, thus supporting the hypothesis that multiple pathogenetic mechanisms underlie motor neuron degeneration in dHMN. Further heterogeneity is expected in dHMN as mutations in the known genes only account for approximately 32.5% of patients with dHMN.2–5 We previously reported a novel form of autosomal recessive dHMN; the ‘Jerash type’ (HMNJ) affecting seven consanguineous families in the Jerash region of Jordan.6 7
SIGMAR1, encoded by the SIGMAR1 gene, is a highly conserved transmembrane endoplasmic reticulum (ER) protein,8 ubiquitously expressed in human tissues and notably enriched in motor neurons.9 SIGMAR1 mostly localises at the mitochondria-associated ER membranes (MAM) domain, where it functions as a molecular chaperone in the ER by stabilising various proteins at MAM and by participating in the degradation of misfolded proteins preventing their accumulation.10–12 Overall, SIGMAR1 is implicated in numerous cellular processes such as ion channel modulation, protein and lipid transport, ER stress response and autophagy, while agonists of SIGMAR1 have been shown to possess neuroprotective properties.12–16
Recently, homozygous or compound heterozygous SIGMAR1 mutations have been implicated in the development of dHMN with pyramidal signs as well as juvenile amyotrophic lateral sclerosis (jALS), two conditions sharing several phenotypical features.17–24 Here, we present functional studies in support of a novel SIGMAR1 mutation causing HMNJ, when present in homozygosity.
Methods
Patients
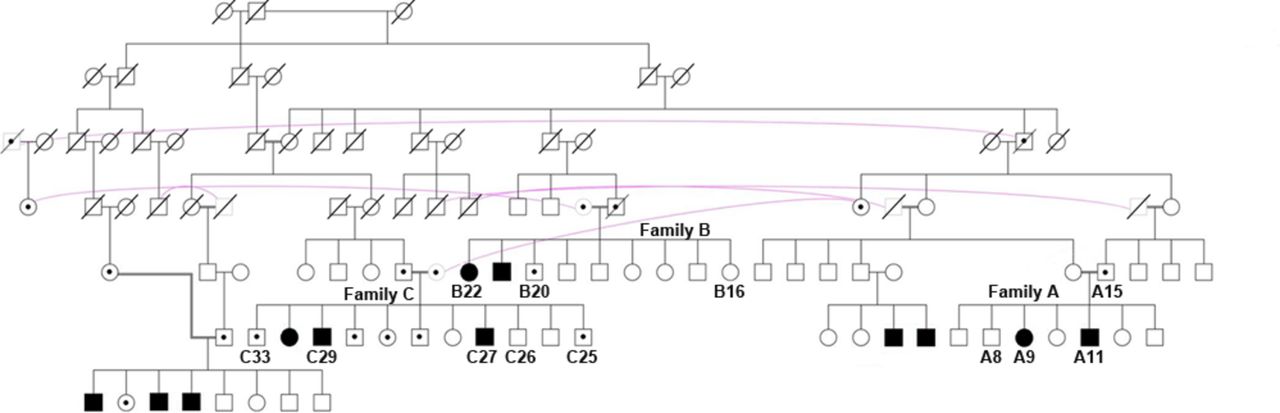
In this study, 12 individuals from 3 consanguineous families with HMNJ were clinically evaluated (figure 1). Five members have progressive distal muscle weakness, while the other seven members are unaffected, some of whom having subjective complaints of pain and discomfort in the feet, on walking. Two of these families were included in our previous report.7 One patient from each previously reported family has been re-examined (B22 and C29) and a new patient (C27) has been ascertained. Two patients from a new family (A9 and A11) are also presented in this report.

Pedigree structure of the three consanguineous families (A, B and C) clinically evaluated for this study. Patients with HMNJ are shown as filled symbols and are homozygous for SIGMAR1N167I . Heterozygous unaffected individuals are depicted with a black dot inside the symbol. Numbered white symbols represent family members that are SIGMAR1WT homozygous, and unnumbered white symbols show family members of unknown genotype. Purple curved lines are drawn between the same individual.
Detailed clinical history, physical and neurological examinations were performed on all 12 cases. Nerve conduction studies were performed to document the sensory and motor compound action potentials and nerve conduction velocities. Median, ulnar, common peroneal and sural nerves were examined. Electromyographic examination was performed to examine the first dorsal interosseous, vastus lateralis and tibialis anterior muscles.
Subjects and samples
The study participants described in the previous report7 were analysed at the Neurogenetics Department of the Cyprus Institute of Neurology and Genetics (CING). The new participants were analysed at the Department of Genetics of Yale University and provided written informed consent to take part in this study. The protocol was approved by the Yale Human Investigation Committee Institutional Review Board (HIC#9512008556). The control cohort for the NGS study was comprised of 2000 previously sequenced samples of an admixture of subjects presenting with early-onset hypertension and normotensives, in an in-house database and subjects found in the NHLBI Exome Sequencing Project database and the Exome Aggregation Consortium V.0.3 database (ExAC).25
Exome sequencing
Genomic DNA from the new HMNJ family was isolated from anticoagulated whole blood using the standard SDS-Proteinase K digestion, Phenol-Chloroform extraction, Ammonium Acetate-Ethanol precipitation method. Genomic DNA from the other 7 HMNJ families as well as from 200 healthy individuals was available from the previous study.7
Exome sequencing was performed by the Yale Center for Genomic Analysis using the exon capture method and the Roche MedExome or Roche V2 capture reagent followed by 75 base paired-end sequencing on the Illumina HiSeq 2000 instrument. Samples were barcoded, captured and sequenced in multiplex, according to the manufacturer’s protocols.
Sequence reads were aligned to the GRCh37/hg19 human reference genome using BWA-MEM local realignment. Quality score recalibration was performed using the GATK pipeline. PCR duplicates were eliminated and variants were called using the Genome Analysis Toolkit Haplotype Caller. Allele frequencies of variants were determined using the ExAC database.25 Pathogenicity of nonsynonymous rare variants was predicted using the Sift and PolyPhen tools.26 27
Mutation analysis
The Neurogenetics Department of CING performed candidate genes analysis and further targeted gene analysis on identification of the candidate mutation with Sanger sequencing. Primers were designed with Primer3 software for the analysis of genes from the candidate region and then for the candidate mutation region. Standard PCR reactions were carried out and PCR products were purified using the QIAquick PCR purification kit (Qiagen, Hilden, Germany), sequenced in both directions with the BigDye Terminator V.1.1 Cycle Sequencing Kit (ABI, California, USA) and then run on the ABI 3130xl Genetic Analyzer. The ABI SeqScape software was used to visualise, align and compare the results of the sequencing runs with the normal sequences of the genes under study. All available DNA samples of family members and control individuals were screened for the identified mutation with the appropriate pair of primers (online supplementary table S1). No other SIGMAR1 variant was detected in the patients’ samples.
Supplemental material
Cell lines
Lymphoblastoid cell lines (LCLs) were previously established from two patients with HMNJ (one male, one female) and three healthy individuals (two males, one female) following written informed consent and were cultured in Roswell Park Memorial Institute medium containing 10% FBS (fetal bovine serum), 2 mM Glutamine, 50 U/mL Penicillin and 50 mg/mL Streptomycin (Biosera, Nuaille, France).
Human SH-SY5Y and HeLa as well as mouse NSC-34 cell lines were cultured in Dulbecco's Modified Eagle's medium supplemented with 10% FBS, 2 mM Glutamine, 50 U/mL Penicillin and 50 mg/mL Streptomycin.
All cell lines were kept at 37°C in 5% CO2.
Expression studies in LCLs
mRNA level
Total RNA was extracted from the aforementioned LCLs of patients with HMNJ and healthy individuals, using the RNeasy Mini Kit (Qiagen). Subsequently, cDNA synthesis was performed with the Protoscript cDNA Synthesis Kit (NEB, Massachusetts, USA). Room temperature (RT)-qPCR was employed on the CFX96 system using the SsoFast EvaGreen Supermix (BioRad, California, USA). Primers specific for SIGMAR1 were used (online supplementary table S1). L19 was employed as a housekeeping gene for this analysis and three independent experiments were performed.
Protein level
Protein extracts were derived from the same LCLs, with the use of a homemade lysis buffer (50 mM Tris-Cl pH 7.5, 150 mM KCl, 2 mM EDTA, 0.5% Triton x-100, 1x Protease Inhibitor Cocktail). The samples were incubated on ice for 1 hour, sonicated three times and centrifuged at 14 000 rpm for 30 min at 4°C. The supernatants were diluted in a final concentration of 1x SDS-PAGE Laemmli sample buffer, boiled for 5 min in 95°C and run in an SDS-PAGE gel. Antibodies to SIGMAR1 and GAPDH (normalisation control) were used for the analysis. Three independent experiments were carried out.
Plasmid constructs and cloning
Whole wild-type and mutant SIGMAR1-ORFs were distinctively amplified, with the use of appropriate restriction sites containing primers (online supplementary table S1), from cDNAs of a homozygous healthy individual and a patient with HMNJ, respectively and cloned into pEGFP-C3 (BD, California, USA) and pFLAG-CMV-2 (Sigma-Aldrich, Missouri, USA) vectors. With the employment of a Flag-tag containing forward primer and an appropriate reverse primer, Flag-tagged SIGMAR1 whole-ORFs were cloned from pFLAG-CMV-2 to the bicistronic vector pIRES2-EGFP (BD). All of the plasmid constructs were sequenced and verified to be free of any nucleotide changes in the SIGMAR1 cDNA.
Expression studies in transiently transfected cell lines
SH-SY5Y and NSC-34 cells were transfected with pIRES2-EGFP-Flag-SIGMAR1 constructs so that Flag-SIGMAR1 and GFP would get cotransfected into the same cells, but expressed as two separate proteins from the same mRNA. Cells were treated with 10 µM MG132 or DMSO (Sigma-Aldrich) for 6 hours. Flag-SIGMAR1 protein’s expression was detected using an anti-Flag antibody. RT-qPCR was also conducted to relatively quantify the mRNA levels, using primers amplifying the exogenous Flag-SIGMAR1 but not the endogenous SIGMAR1. GFP protein and mRNA levels, respectively, were used for normalisation. Three independent experiments were conducted.
Immunoblot analysis
Protein electrophoresis was conducted on a Mini-PROTEAN system, and samples were transferred in a polyvinylidene difluoride membrane on a Mini Trans-blot cell (Biorad). Membranes were blocked for 1 hour in 5% non-fat dry milk in phosphate buffered saline (PBS)/Tween, followed by incubation with the appropriate primary antibody overnight. The next day membranes were washed 3×10 min in PBS/Tween and incubated with the appropriate secondary antibody for 1 hour. After three washes, proteins signals were visualised using the LumiSensor Chemiluminescent HRP Substrate Kit (Genscript, New Jersey, USA), in a UVP BioSpectrum Imaging System (UVP, California, USA). Data were analysed using ImageJ 1.51j8 software. Two-tailed t test was used to assess the p value.
Immunofluorescence
SH-SY5Y cells were grown on coverslips and 24 hours later got transiently co-transfected with the pEGFP-C3 SIGMAR1 and the ER marker mch-Sec61 beta constructs with the use of Lipofectamine 3000 (ThermoFisher, Massachusetts, USA) following the manufacturer’s instructions. Plasmid mCh-Sec61 beta was a gift of Gia Voeltz.28 After 48 hours, cells were treated with 10 µM MG132 or DMSO for 6 hours and then fixed for 10 min at RT with 4% w/v paraformaldehyde in PBS, permeabilised with Methanol for 10 min at −20°C and quenched for 10 min at RT with 50 mM NH4Cl in PBS. Cell nuclei were stained with Hoechst 33 342 and coverslips were mounted with Dako Fluorescent Mounting Medium (Agilent, California, USA). Images were captured with a Zeiss fluorescent microscope using the Axiovision software. Colocalisation index was quantified by Manders’ coefficient using Coloc2 plugin for ImageJ, and two-tailed t test was used to assess the p value.
Antibodies
Goat anti-SIGMAR1, mouse anti-GAPDH (Santa Cruz, Texas, USA), rabbit anti-GFP (Proteintech, Illinois, USA) and mouse anti-FLAG (Sigma-Aldrich) primary antibodies were used. Donkey anti-goat IgG HRP, donkey anti-rabbit IgG HRP (Santa Cruz) and goat anti-mouse IgG HRP (Millipore, Massachusetts, USA) were used as secondary antibodies.
Terminal deoxynucleotidyl transferase dUTP Nick-End labelling (TUNEL) assay
SH-SY5Y and HeLa cells were grown on coverslips placed in 6-well plates and 24 hours later got transiently transfected with pEGFP-C3 SIGMAR1 constructs. Forty-eight hours later cells were fixed for 10 min with 4% w/v paraformaldehyde and permeabilised for 20 min in 0.25% Triton-X. TUNEL assay was conducted using the Click-iT Plus TUNEL Assay for In Situ Apoptosis Detection kit (ThermoFisher). Cell nuclei were stained with Hoechst 33342; coverslips were mounted with Dako Mounting Medium and visualised on a Zeiss fluorescent microscope. The TUNEL-positive nuclei percentage of successfully transfected cells was determined through microscopic analysis of four independent experiments for SH-SY5Y and three for HeLa cells. Approximately 1000 cells were counted for each experiment. Two-tailed t test was used to assess the p value.
Data availability
Data supporting the outcomes of this manuscript and not published within this article are available in an anonymised fashion to qualified investigators on request by directly contacting the corresponding author.
Results
Patient evaluation
This study included clinical evaluation of 12 individuals from 3 consanguineous HMNJ families (figure 1): 3 females and 9 males, with ages between 10 and 58 years.
The five patients presented with progressive, symmetrical lower limb distal muscle wasting and weakness at an early age. Ankle and toe dorsiflexors were more affected than plantar flexors and evertors more than invertors. All patients had pes cavus and all walked with a high-stepping gait. Upper limb involvement were first noted in puberty with the exception of one individual who had wrist and finger extensors more affected than the flexors at a much earlier age. Two patients had mild and three had severe distal muscle weakness and wasting of the lower limbs. Proximal muscles, cranial nerves, coordination and sensory systems were intact. Knee jerks were present in all patients and brisk in all except the oldest one. All patients showed decreased or absent ankle jerks and flexor plantar response, except the youngest patient who exhibited normal ankle jerks and extensor plantar response. Pyramidal signs tend to fade away with age. Motor nerve conduction studies showed decreased compound muscle action potential amplitude with normal or slightly reduced conduction velocities in all five patients, while sensory nerve conduction studies were normal. Electromyography showed active denervation in the form of fibrillations and positive sharp waves, chronic neurogenic changes characterised by the presence of large motor units and reduced recruitment pattern on voluntary contraction of tibialis anterior and first dorsal interosseous muscles. Clinical features and neurophysiological findings were reminiscent of our previous findings.7 Furthermore, mild sensory symptoms in the form of numbness of the feet and to a lesser extent of the hands were recorded in three patients (online supplementary table S2).
Seven non-affected individuals from these three HMNJ families were also clinically evaluated. They all had normal nerve conduction studies and electromyography, no signs of muscle weakness or atrophy, normal ankle reflexes, while Babinski reflex was absent. Some of these individuals reported subjective complaints of pain and discomfort in the feet on walking, which does not seem to be relevant, given the age of individuals and the normal neurophysiological findings.
Identification of a novel SIGMAR1 mutation in patients with HMNJ
HMNJ was mapped to chromosome 9p21.1–p12.7 SIGMAR1 is a gene located within this region that was previously implicated in motor neuronopathies.17–23 Exome sequencing (mean coverage depth: 159X) was performed in a patient with HMNJ and identified a homozygous missense substitution in exon 4 of the SIGMAR1 gene (rs766761162, NM_005866.4: c.500A>T, NP_005857.1: p.N167I, coverage depth: 149x, reads G.0, C.149). No variants in any other genes previously associated with dHMN or other related neuropathies were identified. Furthermore, Sanger sequencing of the 27 patients that were available in the Neurogenetics Department of the CING did not detect any other putative disease-causing variants within the coding region of SIGMAR1. In addition, no C9orf72 repeat expansions were detected in patients with HMNJ tested (data not shown), thus excluding the possibility of a cosegregating C9orf72 expansion.29
Sanger sequencing revealed the presence of the SIGMAR1 variant in a homozygous state, in all 30 affected individuals of the 8 HMNJ consanguineous families. All unaffected parents were identified to be heterozygous while the remaining healthy individuals of the HMNJ families tested, had a single or no copy of the variant (figure 2A). The variant was absent in 100 unrelated healthy control individuals tested. According to ExAC,25 this is an extremely rare variant that has been identified before in a heterozygous state in only one person out of 60 638 worldwide. This SIGMAR1 gene missense mutation causes a single aminoacid substitution (p.Asn167Ile), in which a polar amino acid (Asparagine) is replaced by a hydrophobic amino acid (Isoleucine) within the cytoplasmic domain of the protein (figure 2C), according to UniProtKB.30 The substituted amino acid is well conserved among mammals, but it is not found in other vertebrate classes (figure 2B). Moreover, the identified mutation is predicted to be deleterious by several in silico analysis algorithms, such as SIFT (score 0.02: damaging) and PolyPhen (score 0.984: probably damaging).26 27
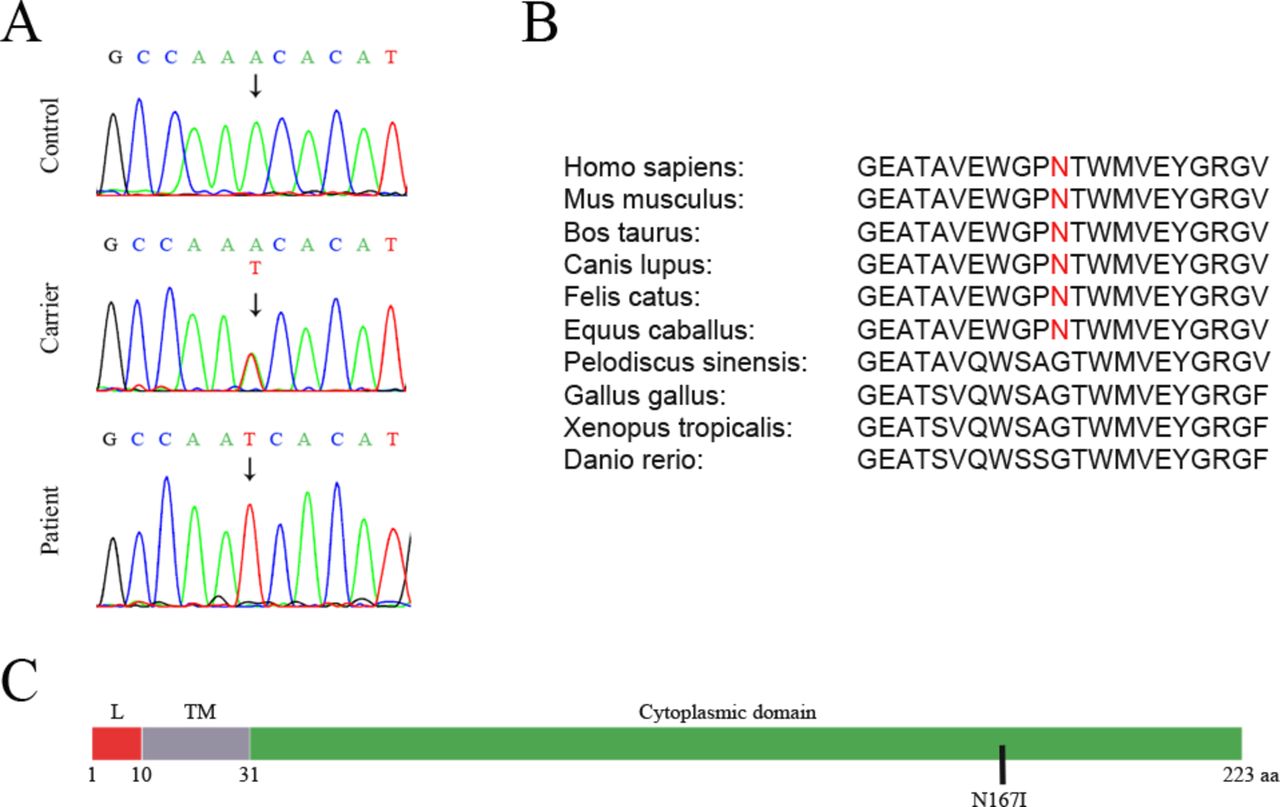
Identification of a homozygous missense mutation (SIGMAR1N167I ) in patients with HMNJ. (A) Sequences demonstrating the identified mutation in genomic DNA from healthy homozygous individuals (control), their unaffected heterozygous parents (carrier) and affected individuals (patient). (B) The affected amino acid is conserved in various mammal species, but is not present in other vertebrate classes. (C) N167I is located in the cytoplasmic domain of the SIGMAR1 protein. L=Luminal domain. TM=Transmembrane domain. Source: UniprotKB.30
HMNJ SIGMAR1N167I aberrant expression and localisation
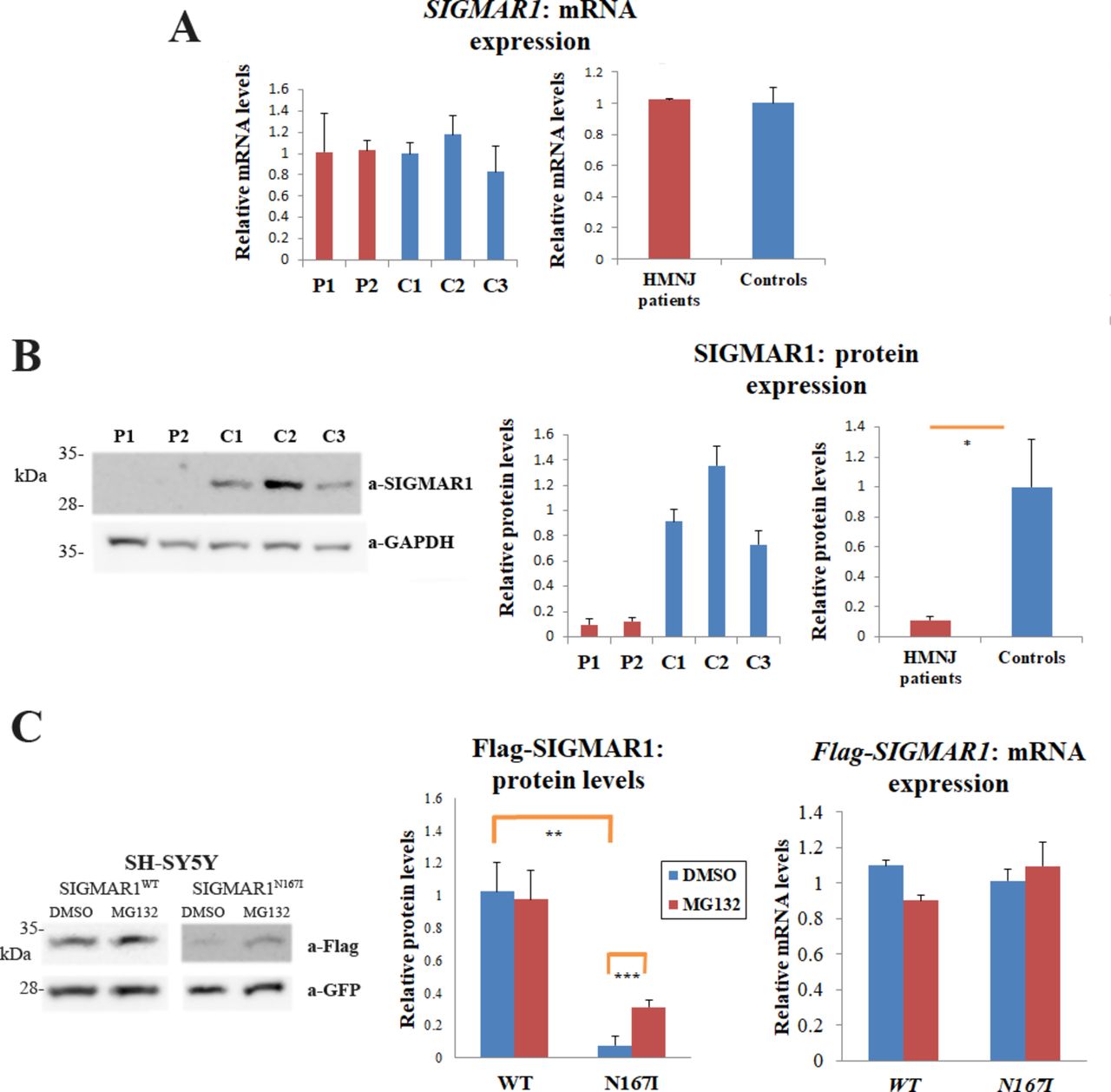
To compare the endogenous expression between SIGMAR1WT and SIGMAR1N167I , we used mRNA and protein extracts from LCLs derived from blood of healthy individuals and patients with HMNJ. SDS-PAGE and Western-blot (WB) analysis revealed that SIGMAR1N167I protein levels are reduced to 11.17%±2.1% (SD) in comparison to SIGMAR1WT (p=0.03) (figure 3B). Interestingly, RT-qPCR showed no significant differences between the mRNA expression of SIGMAR1WT and SIGMAR1N167I (figure 3A), indicating that the reduction in the expression levels is occurring at the protein level.

N167I mutation causes reduction in SIGMAR1’s expression levels. (A) qPCR revealed that SIGMAR1 mRNA expression in LCLs is similar in patients and healthy individuals. The graphs show the relative SIGMAR1 mRNA levels of patients with HMNJ and controls (individually and grouped), after normalisation with the ribosomal protein L19 gene. Error bars depict the SE of the mean (SEM) of three independent triplicate experiments. (B) WB showing that the endogenous protein levels of the SIGMAR1 mutated form from lymphoblastoid cells of patients with HMNJ are reduced in comparison to the control lymphoblastoid samples from healthy individuals, expressing the wild-type SIGMAR1. The graphs show the relative SIGMAR1 protein levels of patients with HMNJ and controls (individually and grouped), after normalisation with the GAPDH protein levels, derived from densitometric quantification of the SIGMAR1 and the GAPDH proteins expression. Error bars depict the SD of three independent experiments. (C) The employment of a GFP-bicistronic vector system reveals a significant drop in protein levels in the case of the heterologous Flag-SIGMAR1N167I in comparison to Flag-SIGMAR1WT, in SH-SY5Y (p=0.001890994) cells. In the presence of MG132, Flag-SIGMAR1N167I protein levels were elevated (p=0.000750874), demonstrating that the mutant SIGMAR1 undergoes ubiquitin-mediated degradation. qPCR showed no significant differences between the wild-type and the mutant heterologous mRNAs, in basal conditions or after MG132 treatment. Error bars depict the SD in the proteins’ expression graph, and the SE of the mean (SEM) in the mRNA expression graph, of three independent experiments. GFP protein and mRNA levels were used for normalisation.
To verify this finding, we transiently transfected pIRES2-EGFP constructs expressing (1) Flag-SIGMAR1WT and GFP and (2) Flag-SIGMAR1N167I and GFP and measured the protein levels of the exogenous Flag-SIGMAR1s. GFP levels were used for normalisation. The expression of Flag-SIGMAR1N167I was almost diminished in comparison to Flag-SIGMAR1WT expression in the human SH-SY5Y cell line (13,41-fold reduction, p=0.0019). Less but still significant reduction was observed in mouse NSC-34 cells (2.2-fold reduction, p=0.0004). Given that the endogenous reduction in expression is occurring at the protein level, we compared the protein expression of the two forms in relation to treatment with the proteasome inhibitor MG132. In the presence of MG132, the exogenous levels of Flag-SIGMAR1N67I were elevated compared with basal conditions, demonstrating that SIGMAR1N167I is degraded through the ubiquitin-proteasome pathway (figure 3C). In SH-SY5Y cells, the expression of Flag-SIGMAR1N167I showed a 4.06-fold increase (p=0.00075) after MG132 treatment, while in NSC-34 cells the expression of Flag-SIGMAR1N167I displayed a 1.99-fold increase after MG132 treatment. RT-qPCR showed no significant differences between the mRNA expression of the two isoforms, under basal or proteasome inhibition conditions. SH-SY5Y data are presented in figure 3C, while NSC-34 data can be found in online supplementary figure S1.
Supplemental material
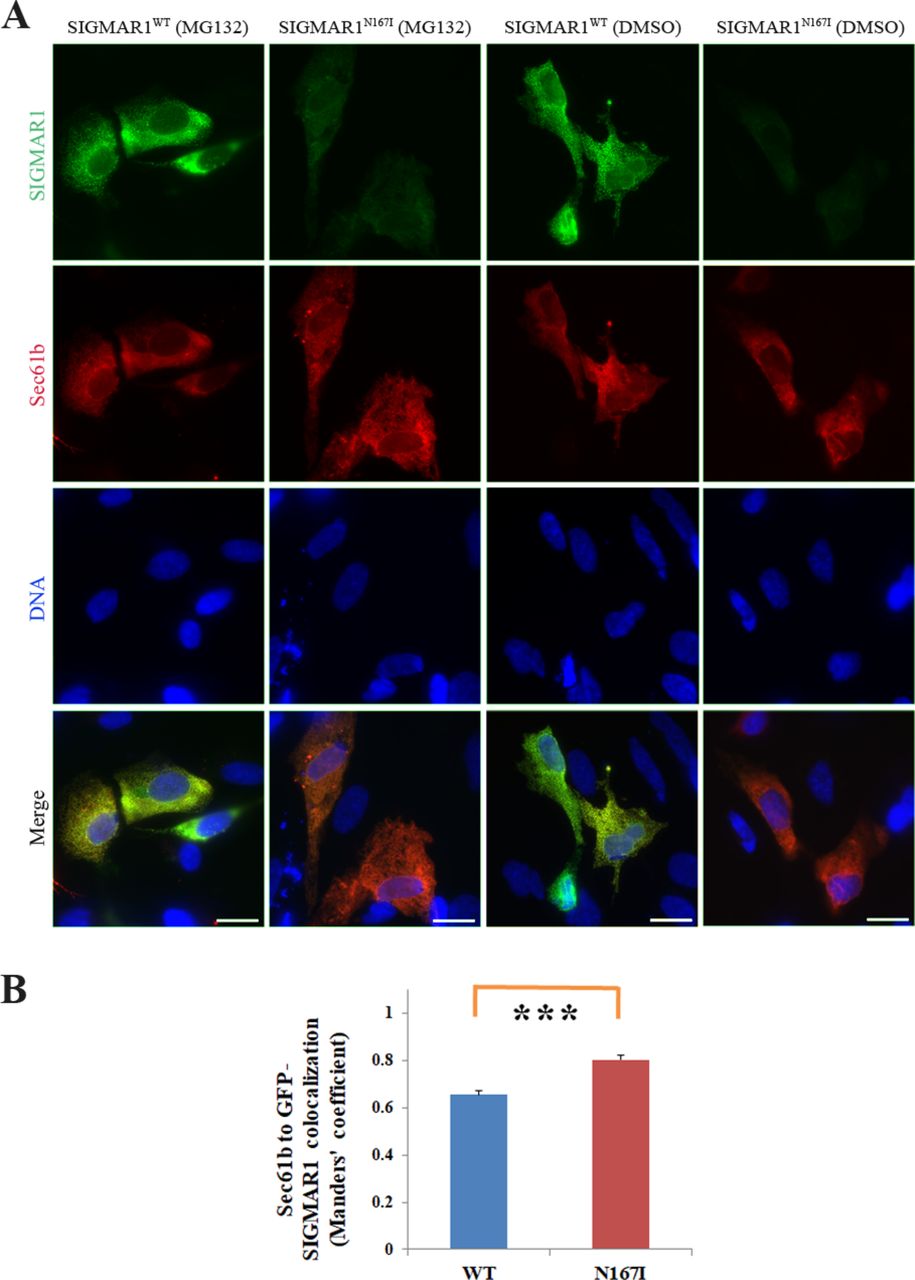
Further evidence of the reduction in the expression of SIGMAR1N167I is given through immunofluorescence experiments. GFP-SIGMAR1N167I fluorescence levels were lower in comparison to GFP-SIGMAR1 in SH-SY5Y cells. As in the case of Flag-SIGMAR1N167I, when the cells expressing GFP-SIGMAR1N167I were treated with MG132, SIGMAR1 levels were raised (figure 4).

Intracellular localisation of GFP-SIGMAR1WT and GFP-SIGMAR1N167I. (A) SIGMAR1WT and SIGMAR1N167I (green) retain colocalisation with endoplasmic reticulum marker Sec61b (red), in the presence or absence of proteasomal inhibitor MG132. Blue colour represents the nuclei. SIGMAR1N167I is showing a lower expression level that is raised in the presence of MG132, consistent with the WB findings of endogenous and exogenous SIGMAR1s in figure 2. Green colour’s exposure is identical in every condition to depict the SIGMAR1 expression differences between them. Scale bars 20 µm. (B) Graph displaying Manders’ coefficient quantification (fraction of Sec61b colocalising with GFP-SIGMAR1 wild-type or mutant) calculated from at least 40 fluorescence microscopy images for each isoform in basal conditions. Error bars depict the SEM of three independent experiments.
Immunofluorescence also showed that even though GFP-SIGMAR1N167I retains its localisation to the ER, it exhibits a more diffused distribution in comparison to the granular allocation of GFP-SIGMAR1WT in SH-SY5Y cells. Manders’ overlap coefficient analysis showed that SIGMAR1N167I colocalises with Sec61b at a higher degree than SIGMAR1WT (p<0001) (figures 4 and 5A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
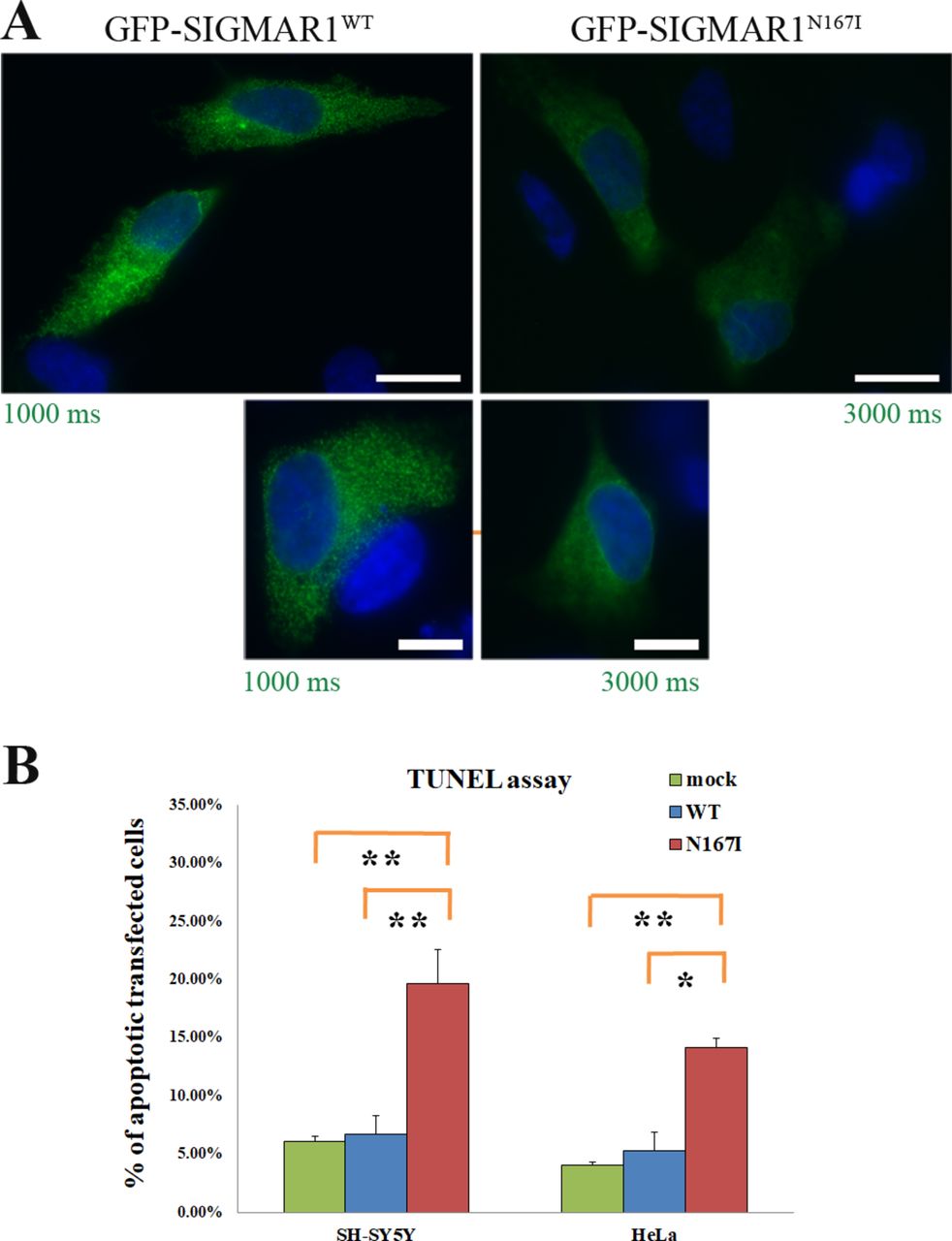
Exogenous GFP-SIGMAR1HMNJ expression consequences in the protein’s localisation and cell viability: (A) SIGMAR1N167I is distributed in a more diffused fashion in comparison to the granular localisation of SIGMAR1WT. Green colour’s exposure is three times higher in the case of SIGMAR1N167I. Scale bars 10 µm in the upper and 5 µm in the lower images. (B) GFP-SIGMAR1N167I’s expression induces apoptosis in SH-SY5Y (p=0.006) and HeLa cells (p=0.023), in comparison to GFP-SIGMAR1WT expression. Additionally, GFP-SIGMAR1N167I’s expression induces apoptosis in SH-SY5Y (p=0.005) and HeLa cells (p=0.002), in comparison to mock-transfected cells as well. No significant differences were found between the mock-transfected and the cells expressing GFP-SIGMAR1WT. Error bars depict the SD of four independent experiments for SH-SY5Y and three for HeLa cells.
SH-SY5Y cells expressing GFP-SIGMAR1N167I show induced apoptosis
Following our observation that GFP-SIGMAR1N167I is degraded through the ubiquitin-proteasome pathway, we proceeded to assess whether its overexpression would cause apoptosis. We employed the TUNEL assay to count the percentage of apoptotic nuclei in transiently transfected SH-SY5Y cells with (1) GFP-SIGMAR1WT , (2) GFP-SIGMAR1N167I and (3) mock cells. The measurements unravelled a statistically significant increase of apoptosis in the GFP-SIGMAR1N167I transfected cells in comparison to the GFP-SIGMAR1WT expressing cells under physiological conditions (with 19.65±2.93 vs 6.70±1.58% SD, p=0.006233) and also in comparison to mock (with 19.65±2.93 vs 6.13%±0.78%, p=0.005123). For validation purposes the experiment was repeated in HeLa cells with similar results (with 14.13±0.81 in GFP-SIGMAR1N167I , 5.33%±1.55% in GFP-SIGMAR1WT and 4.04%±0.28% in mock cells, p=0.022825 for N167I vs WT, p=0.001634 for N167I vs mock). SIGMAR1WT overexpression did not have any effect on apoptosis in basal conditions (figure 5B).
Discussion
We have identified a homozygous SIGMAR1 c.500A>T (N167I) missense mutation cosegregating with HMNJ; an extremely rare variant, based on data from several online browsers and tools. Our findings suggest a loss-of-function effect. The aminoacid change affects SIGMAR1 isoform mSIG-1A, which is the sole brain-specific isoform out of seven in mice.31
Our study included individuals of one newly diagnosed HMNJ pedigree, in addition to individuals from two previously described HMNJ families. The main clinical features such as distal weakness, wasting and the pyramidal signs fading with age were similar to that of our previous report as well as to the other SIGMAR1-associated dHMNs.7 18 20–24 Furthermore, nerve conduction studies and electromyography findings were also comparable to those observed in other SIGMAR1-associated dHMNs and in our previous HMNJ report.7 18 20 21 23 Spasticity in the lower limbs, present in the Omani and the British kindred,22 23 was also present in the patients with HMNJ, unlike the other SIGMAR1-associated dHMN cases.
In recent years, various SIGMAR1 mutations were found to be associated with the development of dHMN with or without spasticity and jALS; however the number of identified patients was low in every SIGMAR1 neuropathogenic mutation with maximum 4 patients with dHMN associated to c.151+1G>T and 6 patients with jALS to c.304G>C.17–24 We report for the first time a SIGMAR1 mutation associated with a neurodegenerative disease in a large number of patients. The SIGMAR1-induced neuronopathies display autosomal-recessive inheritance and exhibit common symptoms like onset in the first or second decade of the patients’ life, progressive weakness and wasting initially of the lower and then of the upper limbs and absence of any sensory abnormalities. The phenotypic spectrum of verified SIGMAR1-induced neuronopathies is presented in table 1.7 17–24 Also recently, compound heterozygous SIGMAR1 mutations were reported in a patient with oldest-old onset ALS.32 However, confirmation of pathogenicity of these mutations with functional studies has not been reported.
Genetic and clinical characteristics of SIGMAR1-associated neuropathies
The finding of a plethora of SIGMAR1 mutations in disease nosology is not unusual; indeed, several mutations reported in other molecular chaperones such as HSPB1 and HSPB8 cause a variety of neuropathies like Charcot-Marie-Tooth Disease, dHMN and ALS.3 4 33–37 Nevertheless, dHMNs with prominent pyramidal signs are considered as forms of jALS, because of the phenotypic similarities they share with these conditions, especially with the autosomal dominant form known as ALS4.38–40 The cellular phenotypes that SIGMAR1N167I is causing, such as reduction of SIGMAR1 protein expression, abnormal localisation and elevated apoptosis, have been observed before in studies of other SIGMAR1 mutations associated with dHMN and jALS.17–19 21 41
Based on our findings of mRNA levels not significantly reduced and mutant protein levels increased in the presence of a proteasomal inhibitor, we propose that the drop in SIGMAR1N167I expression levels is probably due to misfolding, which leads to the proteasomal degradation of the protein. This trimming in SIGMAR1 protein levels was also observed in other possibly misfolded pathogenic SIGMAR1 mutants like the E102Q, L95fs,19 and 31_50del18 causing jALS and dHMN, respectively. It is thus critical for the endogenous SIGMAR1 protein levels to be maintained above an appropriate threshold.
The observed diffused subcellular localisation of SIGMAR1N167I has also been reported for the SIGMAR1 31_50del,41 E138Q and E150K21 mutants causing dHMN, while the jALS-causing E102Q mutant localisation is also aberrant since it forms large puncta.17 41 All these mutations likely disrupt the physiological localisation of SIGMAR1 at the MAM. It is likely that due to SIGMAR1N167I misfolding, the tertiary structure is affected and the mutant has reduced capability of attaching to the MAM, thus remaining cytoplasmically diffused. On the other hand, a possibility could be that the trimeric form of SIGMAR142 is disrupted because of the aminoacid replacement, resulting in the dispersed localisation state that SIGMAR1N167I is found, rather than the granular and compact pattern of the SIGMAR1WT.
Furthermore, SIGMAR1 pathogenic mutants have in common the increase in apoptosis levels. Besides the HMNJ causing N167I mutant that we showed to elevate cell death, the same has been reported for other dHMN-causing mutants such as 31_50del,18 E138Q and E150K,21 as well as for the jALS-causing E102Q mutant.17 43 Therefore, SIGMAR1 is important for cell survival. In addition, even though these mutations are pathogenic only in homozygosity, in the cell lines tested they increased apoptosis in the presence of the endogenous wild-type SIGMAR1. Therefore, when these mutants are overexpressed, they may antagonise with the WT in terms of binding and/or protein-protein interactions.
Reports from in vitro experiments indicate that SIGMAR1 knockdown causes calcium homeostasis defects, ER stress activation and thus apoptosis enhancement.10 14 44 Knockout mice exhibit motor neuron axonal degeneration and death and muscle weakness leading to locomotor deficiency. This is due to MAM disruption, impaired Ca2+ signalling and ER stress activation. Despite the motor defects, the SIGMAR1 knockout mice remain viable, fertile and ambulant. No abnormalities were detected in the sensory neurons, highlighting the specificity and necessity for SIGMAR1’s function in motor neurons.45 46 The benign course of SIGMAR1 knockout mice and the presence of motor but not sensory features resemble HMNJ.
Moreover, the c.238C>T SIGMAR1 truncating mutation (p.Gln80*), which totally diminishes SIGMAR1 protein expression, was identified as the causative factor for dHMN with pyramidal signs in an Omani consanguineous family that is very similar phenotypically with the HMNJ.23 This constitutes a strong indication that SIGMAR1 reduction may be the underlying reason for dHMN development.
We consider the HMNJ phenotype to develop due to a SIGMAR1 loss of function, since endogenous and exogenous SIGMAR1N167I expression is diminished. SIGMAR1N167I homozygosity may leave cells without an efficient SIGMAR1 concentration to carry its subcellular tasks, thus leading to HMNJ.
In conclusion, our findings add further evidence to suggest that SIGMAR1 mutations are implicated in the aetiology of dHMN with mild pyramidal signs and jALS, as also supported by others.17–24 In the light of such compelling evidence, as previously proposed,20 23 it would be advisable that SIGMAR1 mutations are included in diagnostic panels for dHMNs, in the interest of patients with these conditions, as well as for the design of prevention strategies, in the context of public health.
Acknowledgments
The authors thank the patients and their family members for participating in this study.
References
Footnotes
AV and RD contributed equally.
Contributors All authors contributed to the accrual of subjects and/or data. AV, RD, AAAQ and KC contributed to the conception and design of the study and drafted the manuscript. All authors have revised the manuscript for important intellectual content and approved the final version.
Funding This study was supported by the Muscular Dystrophy Association of the United States (Christodoulou K), the Cyprus Telethon (Christodoulou K) and Howard Hughes Medical Investigator (HHMI) funding (Lifton R).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Yale Human Investigation Committee Institutional Review Board (HIC#9512008556).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request.