Article Text

Abstract
FMR1 premutation cytosine-guanine-guanine repeat expansion alleles are relatively common mutations in the general population that are associated with a neurodegenerative disease (fragile X-associated tremor/ataxia syndrome), reproductive health problems and potentially a wide range of additional mental and general health conditions that are not yet well-characterised. The International Fragile X Premutation Registry (IFXPR) was developed to facilitate and encourage research to better understand the FMR1 premutation and its impact on human health, to facilitate clinical trial readiness by identifying and characterising diverse cohorts of individuals interested in study participation, and to build community and collaboration among carriers, family members, researchers and clinicians around the world. Here, we describe the development and content of the IFXPR, characterise its first 747 registrants from 32 countries and invite investigators to apply for recruitment support for their project(s). With larger numbers, increased diversity and potentially the future clinical characterisation of registrants, the IFXPR will contribute to a more comprehensive and accurate understanding of the fragile X premutation in human health and support treatment studies.
- women's health
- reproductive health
- psychiatry
- neurodegenerative diseases
- movement disorders
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The FMR1 gene, located on the long arm of the X chromosome, contains a trinucleotide expansion (cytosine-guanine-guanine (CGG)), normally ranging from 5 to about 40 repeats. In successive generations, the CGG repeat may expand to over 200, causing methylation, gene silencing and absence of the gene’s product, the FMR1 protein (FMRP). The loss or reduction of FMRP is the cause of fragile X syndrome (FXS), the most common inherited cause of intellectual disability and autism. Individuals with 55–200 CGG repeats are referred to as premutation carriers (PC). Premutation-sized FMR1 alleles are relatively common, with a frequency of about 1 in 300 females and 1 in 850 males.1 Both male and female PC are at increased risk for a late-onset neurodegenerative disease—the fragile X-associated tremor/ataxia syndrome (FXTAS),2 and females are at risk for primary ovarian insufficiency (FXPOI),3 affecting reproductive health and family building plans. FXTAS is characterised by intention tremor, cerebellar gait ataxia, peripheral neuropathy, parkinsonism and cognitive decline, and is usually accompanied by brain white matter changes, ubiquitin-positive intranuclear inclusions and atrophy.4 5 FXTAS occurs in an estimated 8%–16% of female PC and 40%–50% of male PC.2 Among female PC, about 20% will experience FXPOI,3 compared with about 1% of women in the general population. The most immediate and significant consequence of FXPOI is reduced fertility as a result of diminished ovarian function. Diminished ovarian function leads to early symptoms of menopause and subfertility, and to reduced responsiveness to fertility treatment. Other health consequences of FXPOI result from early oestrogen deficiency, contributing to low bone density, earlier onset osteoporosis and bone fractures, impaired endothelial function, earlier onset of coronary heart disease and increased cardiovascular mortality and overall mortality.6 The premutation has been associated with a broad range of other clinical problems such as neuropathic pain, fibromyalgia, autonomic dysfunction, hypothyroidism and autoimmune disorders, executive dysfunction and psychiatric problems, referred to as fragile X-associated neuropsychiatric disorders (FXAND)7 or fragile X premutation associated conditions (FXPAC).8
Mechanisms and possible treatments
Current data support three potential mechanisms of FXTAS pathophysiology: transcriptionally activated cellular stress pathways, RNA-mediated toxicity related to RNA binding protein sequestration and repeat-associated non-AUG initiated (RAN) translation of the CGG repeat that generates toxic homopolymeric proteins—the most prominent of which is a polyglycine containing protein called FMRpolyG.9 10 All these proximal events in pathogenesis trigger various cellular cascades that contribute to neurodegeneration, with emerging data suggesting that mitochondrial dysfunction may be central to disease progression.9 11–13 Multiple therapeutic approaches are currently under development, including attempts to target the CGG repeats for degradation or preclude their interactions with specific proteins, suppressing inflammatory pathways and augmenting mitochondrial function and approaches to selectively suppress RAN translation of the repeats.9 10
The molecular mechanisms underlying FXPOI, like FXTAS, are thought to be driven by either toxic RNA gain-of-function or non-AUG RAN translation contributing to abnormal FMRpolyG.3 6 In the gain-of-function scenario, cells attempt to eliminate excess FMR1 transcripts through ubiquitin-proteasome degradation,14 leading to intranuclear inclusions and secondary structures, and then to RNA-protein aggregates in cells which prevents normal cell function or even potentially causing cell death. Although not proven, it is thought that this pathogenic process contributes to diminished ovarian reserve. Following the potential protein dysregulation mechanism, FMRpolyG can sequester specific proteins required for viable cell function through protein-protein interaction. Indeed, ubiquitin-positive inclusions and FMRpolG stained inclusions are found in women with FXPOI and in the premutation mouse model.15 16 Interestingly, mitochondrial dysfunction is a characteristic of both FXTAS and FXPOI.12 13 Given the high degree of overlap in pathophysiology mechanisms, the potential treatment approaches under development described above may be applicable to both conditions. Prior to clinical trials, biomarkers of ovarian function related to FXPOI need to be evaluated to determine their profile throughout the reproductive life span of PC. Such well-characterised biomarkers may be used to identify women who are eligible for clinical trials and serve as possible outcome measures. The involvement in clinical research studies of women who carry a premutation and, when possible, donation of ovarian tissue and other biological samples, will be essential to bring clinical trials to fruition.
The pathophysiology of FXPAC or FXAND has not been specifically studied, although investigators have suggested similar mechanisms to those described above for FXTAS and FXPOI.7 The many and broad range of conditions and symptoms that may fall under these umbrella terms are common in the general population, and therefore it may be more challenging to confirm causal connections to FMR1-mediated pathophysiology. Treatment for FXPAC/FXAND-related conditions has not been empirically studied and follows general clinical guidelines for the presenting symptoms or disorders, although recommendations for minimising toxin exposure and oxidative stress and increasing antioxidant use and exercise have been clinically recommended.7
The International Fragile X Premutation Registry: rationale and development
The International Fragile X Premutation Registry (IFXPR) was developed to facilitate and encourage research to better understand the FMR1 premutation and its impact on human health, to facilitate clinical trial readiness by identifying and characterising diverse cohorts of individuals interested in study participation and to build community and collaboration among PC, family members, researchers and clinicians. The Registry was created in partnership with the National Fragile X Foundation (NFXF) in the USA, the University of California Davis MIND Institute, and members of an international advisory committee (AC). Currently, adults 18 years and older with the premutation, family members without an FMR1 mutation (as potential ‘controls’), and untested individuals at risk of being a PC by inheritance, are invited to register.
An AC was formed, consisting of four psychologists (DH, AW, MR, JG), four movement disorder neurologists (PKT, DAH, SA, ML), a geneticist (SLS), two general physicians (JC, AMCH) and five members or administrators of international fragile X associations (KL, RM, JDW, JC, HR). Once the domains and specific registry items were determined by the AC, the Registry was constructed within Research Data Capture (REDCap; www.project-redcap.org), following guidelines provided by the US National Center for Advancing Translational Sciences Rare Diseases Registry Programme. IFXPR data are managed and stored within REDCap, housed in a cloud data centre at Amazon Web Services and all web-based data transmitted from the registrant to the database are encrypted.
The individual data provided by registrants cover three domains: (1) ‘essentials’ (consent, basic identifying and contact information, FMR1 allele information (including an option to upload FMR1 DNA test results in pdf format), FXTAS and/or FXPOI diagnosis and future biological sample sharing preferences); (2) demographics (eg, gender and sex, education level and occupation, marital status, race and ethnicity) and (3) health conditions during the past year (general health, neurological and psychiatric conditions, reproductive health and autoimmune disorders).
Individuals wishing to participate in the Registry navigate to the NFXF website, which hosts the IFXPR webpage (https://fragilex.org/our-research/projects/premutation-registry/recruitment-application-for-researchers/), where they can find details of the rationale, an introductory video, frequently asked questions, video instructions and supporting materials. Individuals click on an ‘Enroll Now’ button that redirects them to online informed consent. Next, individuals provide information through REDCap surveys (currently in English and Spanish) on the web using a computer or mobile device (survey documents in online supplemental materials). For those with diminished mental capacity, a legally authorised representative may provide consent and input registrant data.
Supplemental material
IFXPR governance and access
The governance procedure is a mechanism for researchers to request and gain approval to work with the IFXPR coordinate to identify IFXPR registrants for recruitment into institutional review board (IRB)-approved research projects and to disseminate study details. Researchers do not have direct access to the registry data or to the participants. They may, however, request a summary of registrant descriptive statistics and/or numbers of registrants potentially eligible for their study and, if needed, a letter of support demonstrating potential access to the Registry for their project. For recruitment activities, the applicant needs to provide proof of IRB approval and additional information within the application, which is then reviewed by the AC for approval prior to any recruitment support. The application is a web-based form that is submitted online via the NFXF IFXPR webpage (IFXPR Recruitment Application for Researchers %7C National Fragile X Foundation). Criteria for application review are based on principles of ethics, scientific rigour, investigator/study team qualifications and potential impact. If an application is approved, the IFXPR staff will email IRB-approved and AC-approved recruitment flyers to eligible registrants. Registrants who are interested in participation are instructed to contact the applicant researcher directly.
IFXPR registrants to date
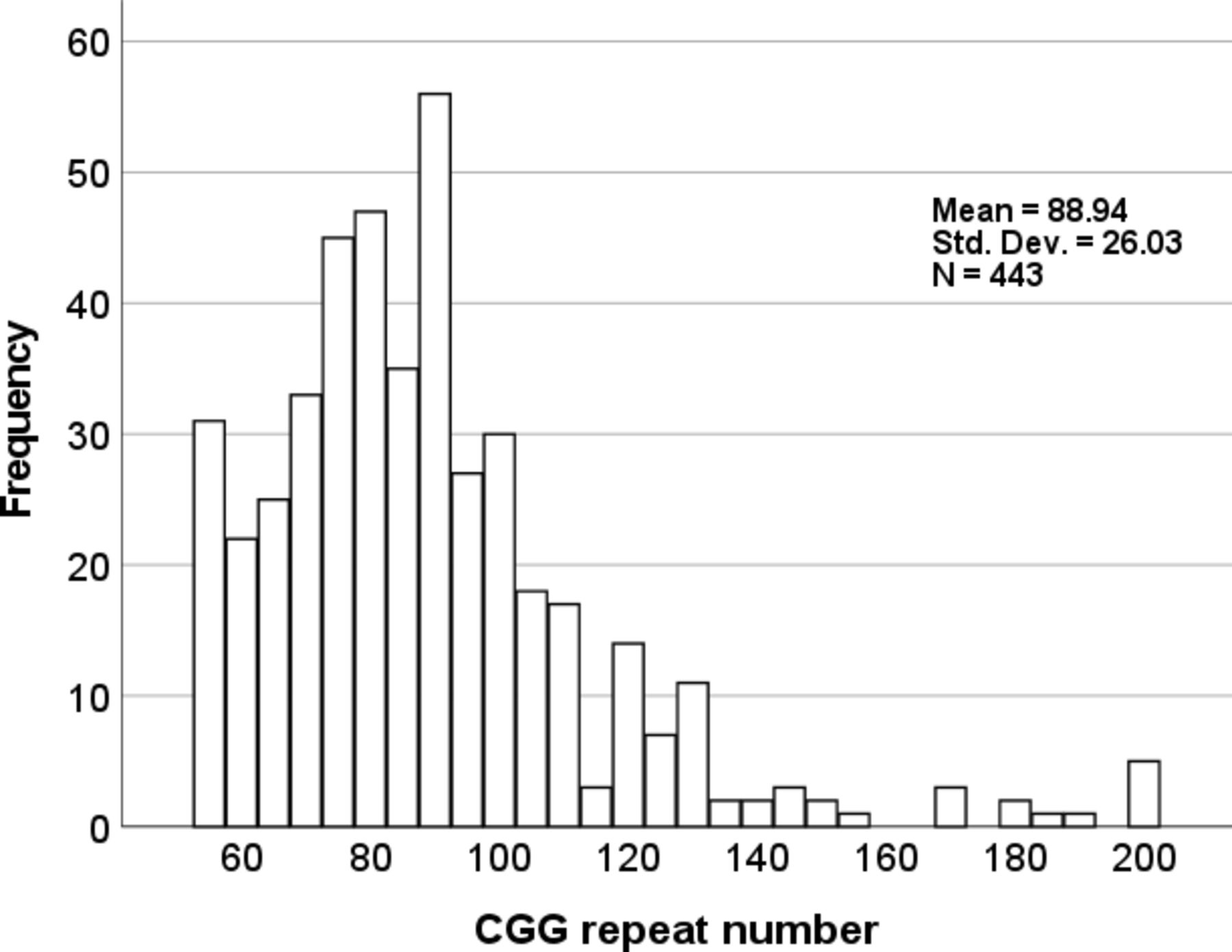
As of January 2022, there were 747 registrants aged 18–90 years (578 with the premutation; 87% female) residing in 32 countries. Note that, although the IFXPR instructions and consent called for PC and family member controls, 14 individuals with intermediate alleles and 14 with full mutation alleles registered. In addition, 116 individuals reported that their FMR1 status is ‘unknown’. Table 1 provides a description of registrants who self-identify as PC (55–200 CGG repeats; n=578). Males range in age from 22 to 81 (n=60; mean=63.3±12.2 SD), and females from 23 to 90 (n=517; mean=48.0±13.2 SD). CGG repeat alleles (the largest is reported when more than one allele size is present) ranged from 55 to 200 (n=443; mean=88.9±26.0 SD; figure 1). The most common reason for FMR1 testing was that ‘a family member tested positive for fragile X’ (67.5%). Only 10.2% of registrants reported that they were tested because of a clinical problem that was thought to be caused by fragile X.
Descriptive statistics and health conditions in the past year among International Fragile X Premutation Registry registrants who self-identify as premutation carriers (as of January 2022)

{kind=link}
Histogram showing the distribution of FMR1 cytosine-guanine-guanine (CGG) repeat expansions in registrants self-identifying as premutation carriers.
Interest in sharing biological samples/tissue
Among all registrants, 77.9% expressed an interest in providing biological samples (20.4% ‘maybe’ or ‘it depends’), and 50.3% were interested in the possibility of being a tissue donor (eg, brain or other body tissues) after death (39.1% ‘maybe’ or ‘it depends’).
Limitations and future directions
There are some important limitations to the registry process and content at present. First, registrant data entry is only possible through use of a computer, smartphone or tablet. This prevents access by individuals in less developed regions who do not have internet access, thereby limiting diversity and inclusion, but it does open access to a large number of carriers who have no ready connection to local researchers and clinicians. A drawback of this registrant-driven process is that local experts are bypassed and do not yet have the opportunity to contribute data. However, two future expansions of the Registry include: (1) clinical characterisation and validation of registrant data through partnerships with fragile X clinics (eg, confirmation of FXTAS diagnosis using standardised protocols) and (2) collection of laboratory data, including FMR1 genomic and molecular measures, as well as other key biomarkers relevant to risk, progression and other disease characteristics. Second, registrants may not have access to accurate information about their health or may misunderstand details of information requested by the registry. As such, at this stage, the accuracy of health details, FMR1 status and other data should be confirmed by researchers. Third, the ethnic and racial diversity of registrants is currently limited. The IFXPR is attempting to facilitate inclusion by employing strategies that could help increase participation, such as building relationships with minority-serving physicians outside of the specialty clinics, to encourage minority patient referrals. Considerable outreach with educational interventions and making clear the possible harms and benefits associated with premutation research are essential to engendering trust and increasing participation among all ethnic/racial communities. Fourth, the proportion of male:female PC registrants is low. Improved outreach through FXS family support groups and neurology specialists is needed to recruit a larger number of males with the premutation, or with a high likelihood of carrying a premutation. Fifth, despite clear messaging that family member controls are encouraged to register, to date the IFXPR only includes a small number of such individuals. Finally, the IFXPR does not presently collect prospective longitudinal data over time, which would constitute a research study. In the future, with appropriate IRB approval, we may obtain registrant consent to collect cumulative data over time which may be useful for charting the natural histories FXTAS, FXPOI and other premutation-associated conditions.
Ethics statements
Patient consent for publication
Ethics approval
The University of California Davis Institutional Review Board exempted this study. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors extend their thanks and appreciation to the registrants of the International Fragile X Premutation Registry for sharing important information to promote and improve research focused on fragile X-associated disorders.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors DH conceived and developed the Registry and authored the manuscript. HR developed and promoted the Registry, developed the webpage, advised on content and reviewed the manuscript. RM led international outreach efforts and reviewed and edited the manuscript. GE is the Registry coordinator. JF assisted with Registry development. KL and JC led Registry promotion efforts in Australia. AMCH directed Spanish translation efforts and assisted with promotion of the Registry in Colombia. SLS, JDW, PKT, DAH, ML, TH, SKS, JG and AW provided critical input on Registry development and reviewed and edited the manuscript.
Funding Funding for development and maintenance of the Registry came from the MIND Institute, the National Fragile X Foundation and a family philanthropic gift.
Competing interests DH is a member of the Clinical and Scientific Advisory Committee and the Clinical Trials Committee of the National Fragile X Foundation.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.