Article Text

Abstract
Introduction Pigmentary mosaicism (PM) manifests by pigmentation anomalies along Blaschko’s lines and represents a clue toward the molecular diagnosis of syndromic intellectual disability (ID). Together with new insights on the role for lysosomal signalling in embryonic stem cell differentiation, mutations in the X-linked transcription factor 3 (TFE3) have recently been reported in five patients. Functional analysis suggested these mutations to result in ectopic nuclear gain of functions.
Materials and methods Subsequent data sharing allowed the clustering of de novo TFE3 variants identified by exome sequencing on DNA extracted from leucocytes in patients referred for syndromic ID with or without PM.
Results We describe the detailed clinical and molecular data of 17 individuals harbouring a de novo TFE3 variant, including the patients that initially allowed reporting TFE3 as a new disease-causing gene. The 12 females and 5 males presented with pigmentation anomalies on Blaschko’s lines, severe ID, epilepsy, storage disorder-like features, growth retardation and recognisable facial dysmorphism. The variant was at a mosaic state in at least two male patients. All variants were missense except one splice variant. Eleven of the 13 variants were localised in exon 4, 2 in exon 3, and 3 were recurrent variants.
Conclusion This series further delineates the specific storage disorder-like phenotype with PM ascribed to de novo TFE3 mutation in exons 3 and 4. It confirms the identification of a novel X-linked human condition associated with mosaicism and dysregulation within the mechanistic target of rapamycin (mTOR) pathway, as well as a link between lysosomal signalling and human development.
- TFE3
- intellectual disability
- lysosomal metabolism
- pigmentary mosaicism
- storage disorder
Statistics from Altmetric.com
Introduction
Intellectual disability (ID) affects about 3% of individuals worldwide and raises significant issues in terms of diagnostic, management and genetic counselling. The presence of pigmentation anomalies in a patient with ID represents helpful clinical clues in order to narrow the range of aetiological hypothesis. Hypomelanosis of Ito (HMI, MIM #300337) is an unspecific term encompassing a heterogeneous group of disorders characterised by cutaneous hypopigmented whorls and streaks along Blaschko’s lines and variable extracutaneous features affecting the musculoskeletal and nervous systems.1 The cutaneous pattern therefore represents a non-specific hallmark of mosaicism in these neurocutaneous conditions. Genetic mosaicism is due to postzygotic mutations, either chromosomal rearrangements or point mutations, whereas random X inactivation in females leads to functional mosaicism.2 Unravelling the molecular basis of pigmentary mosaicism (PM) is still a challenge due to clinical and genetic heterogeneity, technical difficulties in detecting mosaic mutations by classical sequencing approaches and the complexities of obtaining affected tissue. As part of a collaborative group, we recently reported de novo mutations in exons 3 and 4 of transcription factor E3 (TFE3) as the cause for HMI in four unrelated individuals, including one male, as well as syndromic ID without pigmentary disorders in a female.3
TFE3 belongs to the MITF family of mammalian basic helix–loop–helix zipper transcription factors, together with TFEB and TFEC; all four can form homodimers or heterodimers with each other.4 Embryonic expression of TFE3 orthologues Tfe3a and Tfe3b was demonstrated in the zebrafish in a wide range of tissues.5 TFE3 subcellular localisation plays a crucial role in the regulation of cellular homeostasis and embryonic stem cell (ESC) differentiation. The phosphorylated TFE3 is retained in the cytoplasm, whereas dephosphorylated protein translocates to the nucleus to promote the transcription of target genes involved in lysosomal biogenesis and autophagy.6 TFE3 relocalisation to the nucleus is driven on various stressors, such as starvation,7 8 DNA damage,9 mitochondrial damage,10 Golgi stress11 and pathogens12 in an mTORC1-dependent manner, and oxidative stress13 or cadmium exposition14 in an mTORC1-independent manner. Moreover, lysosomal signalling-induced nucleocytoplasmic redistribution of TFE3 is essential to the regulation of ESC renewal.3 15 By restricting nuclear localisation and activity of Tfe3, lysosome activity, the tumour suppressor protein folliculin and the Ragulator protein complex enable the exit from pluripotency and therefore drive differentiation. Conversely, enforced nuclear Tfe3 enables ESCs to withstand differentiation.15 In humans, TFE3 mutations have long been known in cancer. Gene fusions by translocations or other chromosomal rearragements involving TFE3 and five partner genes have indeed been reported to occur in a subset of renal cell carcinomas (RCCs), referred to as ‘TFE-fusion RCC’, and, more rarely, to lung sarcoma and perivascular epithelioid cell tumours.16 Beyond these data on TFE3 function, by the report of a series of 17 individuals harbouring de novo mutations in exons 3 and 4 of TFE3, we emphasise their phenotype and bring additional clinical insight toward the recognition of this novel developmental disorder.
Methods
Patients
We clustered 17 patients with de novo TFE3 variants from 13 cohorts using the GeneMatcher tool.17 Patients 1, 2 and 7 were from a research cohort (Mosaïc Undiagnosed Skin Traits And Related Disorders, M.U.S.T.A.R.D.) compiled in the genetics department of Dijon Hospital to identify molecular bases of PM that included 26 individuals. Twelve patients were investigated for diagnostic purposes because of ID (patients 3–5, 8 and 10–17). Patients 6 and 9 had a clinical diagnosis following the identification of the gene; patient 9 previously had whole-exome sequencing (WES) done, which had been considered as negative and was interpreted again to focus on TFE3. We obtained written informed consent for diagnostic or research purposes testing from all subjects or their legal representatives. We extracted genomic DNA from fresh skin, cultured skin fibroblasts and blood samples using the Gentra Puregene Blood and Tissue Extraction Kit (Qiagen). We assessed genomic DNA integrity and quantity by agarose gel electrophoresis, NanoDrop spectrophotometry and Qubit fluorometry (Thermo Fisher).
Whole-exome sequencing
Routine trio WES was applied by all participants according to their own diagnostic protocols and platforms.18 19 Candidate de novo mutational events were identified by focusing on protein-altering and splice-site changes (1) supported by at least three reads and 10% of total reads in the proband; (2) absent in both parents, as defined by variant reads representing less than 5% of total reads; (3) at base-pair positions covered by at least four reads in the entire trio; and (4) present at a frequency less than 1% in the Single Nucleotide Polymorphism database dbSNP (build 147) and 0.1% in the Exome Aggregation Consortium (http://exac.broadinstitute.org/). For patient 6, the general assertion criteria for variant classification are publicly available on the GeneDx ClinVar submission page (http://www.ncbi.nlm.nih.gov/clinvar/submitters/26957/).
TFE3 sequencing
For Patient 6, we amplified regions of interest using custom intronic primers and long-range polymerase chain reactions with the PrimeSTAR GXL DNA Polymerase (Takara Bio, Saint-Germain-en-Laye, France). We pooled, purified, and quantified PCR amplicons from each affected individual. We prepared libraries using the Nextera XT DNA Sample Preparation kit (Illumina, Paris, France), which involves simultaneous fragmentation and tagging of DNA fragments by in vitro transposition. We performed paired-end sequencing reactions of 150 bp reads on a MiSeq platform using 300-cycle reagent kits (v2; Illumina, Paris, France).
Determination of the X-chromosome inactivation (XCI) pattern
XCI studies were performed in four of the seven females, in blood and fibroblasts for patients 1 and 3, in fibroblasts only for patient 2 and in blood only for patient 7.
XCI pattern was estimated at the HUMARA locus as described previously20 on DNA extracted from uncultured fibroblasts. Fluorescent PCR products were analysed by capillary electrophoresis on an ABI3130XL genetic analyser (Applied Biosystems), and peak areas were generated with the GeneMapper software (Applied Biosystems). Skewing was defined as greater than 85% of one X allele active.
Results
We describe a series of 17 patients carrying a de novo mutation in TFE3, 5 of them being previously published with limited clinical information.3 Twelve were females and five were males; their age ranged from 12 months to 22 years. Five were referred for HMI, five for syndromic ID and five for suspicion of storage disorder.
Clinical data
The clinical features are summarised in table 1. Additional information can be found in online supplementary data 1.
Supplemental material
Clinical and molecular features of the 17 patients with an TFE3 mutation
Neonatal course was remarkable for nine patients: history of jaundice, hepatomegaly or feeding difficulties was reported for three patients each, hypoglycaemia for two and cholestasis for one. All these features were transient.
Developmental delay, usually severe and noticeable from the first months of life, was a constant feature in all the individuals. Only 6 patients were able to walk at the time of the study, whereas 11 were still unable to walk. All patients were non-verbal, except for two older patients who could speak a few words. Neurological examination was abnormal in 12 individuals and consisted in truncal hypotonia, associated with lower limb spasticity (6 individuals) or ataxia (2 adults). Behavioural issues such as autistic features and sleeping disturbance were noted for 11 patients. Eleven patients developed epilepsy, onset in the first decade and characterised as intractable in three of them. Brain MRI was normal in 10 individuals and abnormal in 6 patients (hydrocephaly, short corpus callosum, Dandy-Walker malformation, arachnoid cyst, periventricular white matter lesions, delayed myelination and cerebral atrophy). The sensory anomaly was congenital hearing loss (5 patients), and ophtalmological anomalies (10 patients) consisted of strabismus, hyperopia, retinal degeneration, depigmented macule on the iris, oculomotor apraxia or impaired vision of cortical origin.
Facial dysmorphism shared among the patients consisted in coarseness, flat nasal bridge, short nose with anteverted nares, widely spaced eyes, almond-shaped eyes, thick lips, facial hypertrichosis, fleshy earlobe, and full and pink cheeks (figure 1). Twelve patients had pigmentation anomalies, located on Blaschko’s lines for 10 of them (figure 2). One was diagnosed at 4 years old with histiocytofibroma. Moderate to severe postnatal growth retardation affected 10 patients, who had a length between −2.0 and −4.5 SD. Obesity affected 13 individuals. Skeletal anomalies were frequent (11 individuals) and consisted of flat or clubfeet, hyperlordosis, scoliosis, hip dislocation, limitation of elbow extension and genu valgum. Recurrent infections of the upper airways were noted in five patients; one had a documented neutropenia. Early-onset chronic interstitial lung disease was reported in two patients; nail clubbing was noted in two individuals. Visceral malformations consisted of congenital heart defect (left ventricle dilatation, aortic insufficiency and patent ductus arteriosus) in three patients, umbilical hernia in three individuals, lateral semicircular canal dysplasia, posterior plagiocephaly, sleep apnoea syndrome, anteriorly displaced anus and hypospadias in one individual each.

Facial phenotypes of seven patients. (A–C) Patient 5, aged 6 months (A,B) and 3 years (C). (D) Patient 8, aged 5 years. (E,F) Patient 2, aged 5 and 20 years. (G–I) Patient 3, aged 1 year (G) and 3 years (H,I). (J,K) Patient 13, aged 22 years. (L–O) Patient 6, aged 8 months (L), 20 months (M) and 3 years (N,O). (P,Q) Patient 10 aged 22 years.

Cutaneous features. (A) Patient 3: umbilical hernia, widely spaced nipples and hypopigmentation on the left side of the abdomen. (B) Patient 17: hypotonia, umbilical hernia and widely spaced nipples. (C) Patient 8: Blaschko’s lines on the back. (D) Patient 6: hypopigmentation on the left side of the abdomen. (E) Patient 7: Blaschko’s lines on the abdomen and right side of the trunk. (F) Patient 13: hand; note clubbing of thumbnail and loose skin. (G) Patient 17: Blaschko’s lines on the left side of the abdomen. (H) Patient 17: hand; note tapering fingers and wrinkled skin. (I) Patient 1: linear hypopigmentation on the back. (J) Patient 6: Blaschko’s lines on the back. (K) Patient 3: Blachko’s lines on the right lower limb. (L) Patient 6. Blaschko’s lines on the right lower limb. (M,N) Patient 7: linear hyperpigmentation on the lower limbs. (O) Patient 11: Blaschko’s lines on the back.
Molecular results
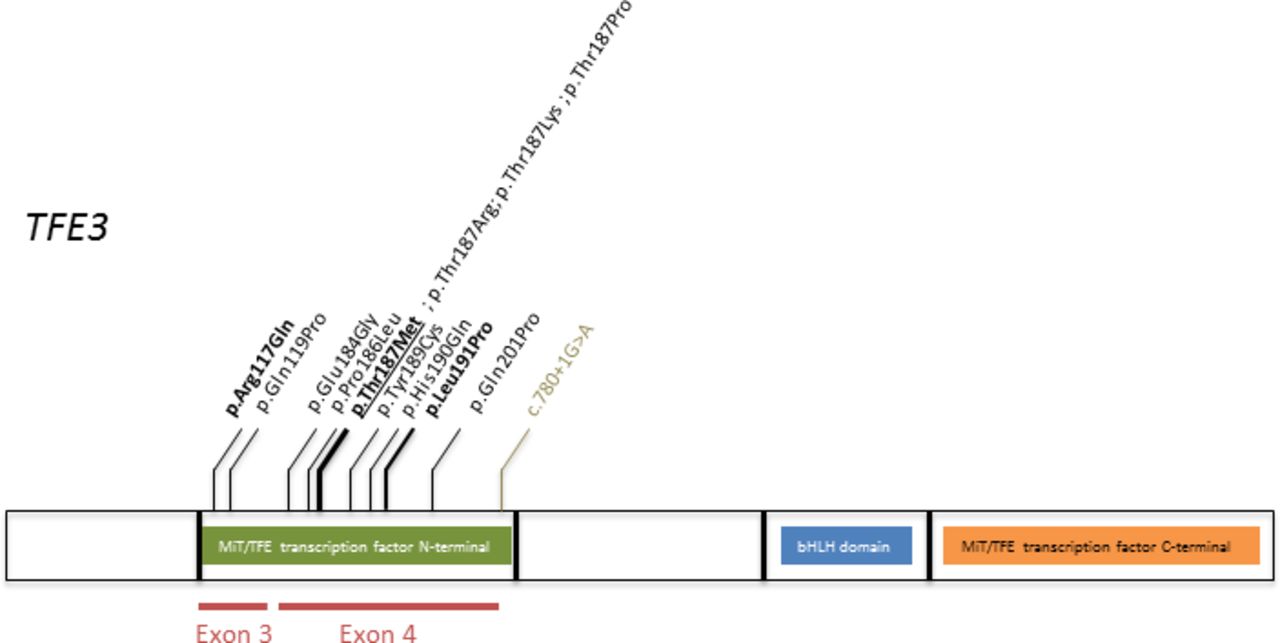
The characteristics of the 13 different de novo TFE3 variants identified in the 17 unrelated individuals are summarised in table 2. All but one were missense variants, affecting nine different aminoacids. One was a splice donor mutation. This mutation was reported a few weeks ago in a patient with a similar phenotype.21 Two variants were localised in exon 3 and 11 in exon 4 (figure 3). All were absent from public databases and were predicted to be pathogenic by prediction softwares.
{kind=link}
{kind=link}
{kind=link}
TFE3 protein and localisation of the missense variants identified. In bold are variants identified in two patients. In bold and underlined is the variant identified in three patients. In green is the intronic variant.
Molecular data of the 13 de novo TFE3 mutations: characteristics, inheritance, frequency in the public database GnomAD, prediction scores regarding pathogenicity (Polyphen, Grantham and CADD (Combined Annotation Dependant Depletion) scores); the transcript is NM_006521.5
The putative mosaicism was assessed through X inactivation studies in females and analysis of the exome sequencing data in males, by checking the total number of reads covering the variant, as well as the number of reads supporting the presence of the variant (table 1). Allele frequencies in females were always consistent with a constitutional heterozygous mutation. X inactivation was skewed in blood of the female patients 1, 3 and 7 and in fibroblasts of Patient 2. X inactivation was random in fibroblasts of Patient 1 and 3. Regarding the male patients, the mutation was identified in 65% of the reads for Patient 17% and 88% of the reads for Patient 15 (106/120). No mosaicism was detected in the blood of Patient 13, 14 and 16 despite the presence of pigmentary manifestations in Patient 16.
Discussion
TFE3 functions in signalling of the mechanistic target of rapamycin (mTOR) complex 1 (mTORC1). The PIK3-AKT-mTOR pathway plays a role in the regulation of cellular growth, proliferation, survival and metabolism. Overactivation of the mTOR signalling is responsible for neurocutaneous disorders and cancers.22 Somatic mutations in TSC1, TSC2, AKT3, PIK3CA and MTOR are responsible for focal cortical dysplasia type II (MIM607341),23–25 hemimegalencephaly26 and megalencephaly.27 The phenotype ascribed to germline TSC1/TSC2, PTEN, MTOR and PK3R2/AKT3/CCND2 mutations – respectively in tuberous sclerosis (TS, MIM 191100), Cowden syndrome (CS, MIM 158350), Smith-Kingsmore syndrome (MIM 616638) and Megalencephaly, Polymicrogyria, Polydactyly and Hydrocephalus syndrome 1, 2 and 3 (MPPH1/2/3, MIM 603387/615939/615938) - is characterised by the association of ID, epilepsy, brain malformations and skin tumours. Similarly, all the individuals harbouring a de novo TFE3 mutation reported in the series presented with a severe neurodevelopmental disorder. Delayed psychomotor development was constant; the youngest patient to acquire independent walking was 30 months old, and more than half of the patients aged over 18 months (57%), did not acquire walk at the last examination. Conversely to patients with MTOR, AKT3 or PTEN mutation, none of the patients described in this series had macrocephaly. Brain imaging was abnormal in 35% of the patients. Hydrocephaly and corpus callosum dysgenesis, identified in respectively three and one individual, were previously reported in patients with mosaic gain of function MTOR mutations.28 29 One patient had surgery to remove an early-onset histiocytofibroma. However, no other skin tumour was reported, either in this patient or in any other from the series.
Pigmentation anomalies, along Blaschko’s lines or, for one patient, as a large hyperpigmented area, were present in a majority of the individuals (71%) in the series, including 40% of the males and 83% of the females. PM along Blaschko’s lines is highly suggestive of genetic mosaicism.30 Genomic mosaicism is defined by the presence of at least two cell populations with different genotypes in an individual originating from one zygote and mainly occurs through post-zygotic event, whereas females can present with functional (epigenetics) mosaicism due to X inactivation.2 PM is a classical feature of X-linked male-lethal genodermatosis, such as incontinentia pigmenti (IP, MIM #308300), focal dermal hypoplasia (FDH, MIM #305600), chondrodysplasia punctuata type 2 (Conradi-Hunermann-Happle syndrome, CDPX2, MIM #302960) and linear skin defects with multiple congenital anomalies (LSDMCA1, MIM #309801). In these conditions, the overwhelming predominance of affected females is a consequence of the male lethality, and the PM a manifestation of the functional mosaicism occurring in females. Similarly, the majority of individuals with de novo TFE3 variants in our cohort were females (sex ratio female:male was 12:5 (2.4)). The study of X-inactivation on non-cultured fibroblasts was consistent with functional mosaicism in two affected females with PM who harboured random X-inactivation, whereas a third female without PM had skewed X-inactivation. In IP, FDH and CDPX2, most hemizygous males die in utero; however, there have been reports of surviving males 31–33 with an estimated prevalence around 10% in FDH and IP.32 34 The majority of them are explained by post-zygotic mutations or chromosomal anomalies (Klinefelter syndrome). Non-mosaic males have also been reported in FDH and IP – respectively about 17% and 45% of the affected males harbour a non-mosaic variant.32 35 In our series, males represented 29% of the patients with a de novo TFE3 variant. A mosaic variant was identified in blood for half of them; none had Klinefelter syndrome. Interestingly, mosaicism was detected in only one out of the two males with PM, and one male with a mosaic variant had no pigmentation anomalies noted on examination. It is still possible that subtle pigmentation anomalies were missed on examination. Moreover, somatic mosaicism can be difficult to detect; recent studies have shown that a large proportion of de novo mutations presumed to be germline had in fact occurred as post-zygotic event.36 In the males of this cohort, WES was performed on leucocytes-derived DNA and no other tissue was studied; therefore, it is possible that a low mosaicism was not detected. Finally, it is probable than TFE3 mutations account for a significant proportion of patients with HMI. Indeed, in this population, the high frequency of ID, epilepsy, coarse facial features has long been emphasised in the literature.37
By its ability to bind the coordinated lysosomal enhancement and regulation (CLEAR) sites in the promotor region of target genes, TFE3 is involved in the control lysosomal biogenesis, autophagy and endocytosis.8 Several patients of the series indeed had clinical and biochemical features that pointed toward an inborn error of metabolism. Lysosomal storage disorder was suspected due to the variable association of coarse facial features (88%), skeletal anomalies (65%) –flat or clubfeet, hyperlordosis, hip dislocation, limitation of elbow extension, genu valgum, scoliosis–, postnatal growth retardation (59%), history of speech or developmental regression (29%) congenital hearing loss (29%), recurrent upper airways infections (29%) neonatal liver anomalies such as hepatomegaly and cholestasis (18%), upper airways infections (24%), umbilical hernia (18%), sleep apnoea syndrome (6%) and aortic insufficiency (6%). Other metabolic anomalies observed in the series were obesity, defined in children by body mass index (BMI) (weight/height2) above the WHO curve, present in the oldest patients (76%), neonatal transient hypoglycaemia (12%), and hyperlactataemia (6%). Dysregulation of lipid metabolism, via suppression of thermogenesis and decreased lipolysis, thus leading to increased adipose tissue, was previously observed in adipose-specific TFE3 transgenic mice.38 Similarly to lysosomes, mitochondrias have a key role in cellular metabolism, including autophagy; recent data demonstrate that mitochondrial and lysosomal metabolisms are interrelated.39 Muscle biopsy, performed in two individuals from this cohort, showed fat and glycogen accumulation, muscular fibre size irregularity, without evidence of mitochondrial dysfunction. Interestingly in the more recent data, evidences showing that TFE3 plays a role in the regulation of the circadian oscillations of the expression of genes involved in autophagy and lipid metabolism, and that Tfe3 knock-out mice had abnormal circadian behaviour.40 Indeed, in our series, five patients (29%) were noted to have sleep disturbance: this could be due to circadian rhythms alteration. Finally, TFE3 has been shown to be involved in the regulation of innate immune response in macrophages via the FLCN-AMPK signalling axis,41 and of B-lymphocytes activation.42 Along these lines, four patients of the series (24%) had a history of recurrent infections, associated with documented neutropenia in one of them. As shown in table 3, a summary of the frequency of the features observed in the cohort, facial dysmorphism was constant and strikingly similar among the patients: more than two-third had anteverted nares, broad flat nasal bridge, almond-shaped and widely spaced eyes, puffy cheeks and coarse facial features (thick lips and fleshy earlobes); more than half had facial hypertrichosis. All individuals presented with at least four of the above features. One patient had an extreme facial phenotype of hypertelorism, bifid nose and bilateral cleft lip and palate. Whether these frontonasal dysplasia features may be associated with the TFE3 mutation remains unclear; no other mutation in known genes was found in Patient 1’s exome sequencing data.
Frequency of the clinical features observed in the series
TFE3 is a highly conserved protein, intolerant to loss of function as supported by data from the GnomAD browser43 (probability of being loss-of-function intolerant (pLI) evaluated at 0.98, observed:expected ratio=0.06) and to missense variants (Z=2.15). Moreover, TFE3 does not, or only in a few tissues, escape X inactivation, suggesting that TFE3 gene dosage is crucial to cell function.44 45 In vitro, Villegas et al recently showed that the absence of either TFE3 exon 3 or 4 resulted in a nuclear gain-of-function Tfe3 allele in ESCs, indicating that both exons 3 and 4 are required for Tfe3 inactivation.3 Nuclear localisation and resistance to differentiation were proved in Tfe3 knock-out (K.O.) ESCs expressing murine Tfe3 alleles (Gln118Pro and Pro185Leu, corresponding to mutations Gln119Pro and Pro186Leu identified in individuals referred to as patients 1 and 2 in this series). Based on the analysis of TFE3 secondary structure,46 indicating that residues 110–215 are predicted to form a domain of two stable alpha helices that might be disrupted by mutations in exons 3 and 4, and the observation of a similar phenotype in patients harbouring mutations in exons 3 and 4, it was suggested that Tfe3 exons 3 and 4 form a Rag binding fold whose structural integrity is indispensable for lysosome-mediated cytoplasmic Tfe3 inactivation.3 In this series, the recurrent mutations Arg117Gln, Leu191Pro and Thr187Met were present in respectively two, two and three patients. The aminoacid in position 187 was mutated in five patients. In addition, 13 of the described mutations were localised between positions p.184 and p.201: this proximity could account for the absence of obvious genotype–phenotype correlation. The canonical splice site variant in intron 4 identified in patient 9 might lead to in-frame exon skipping of exon 4. The clinical picture of the patient with this splice site variant perfectly fits with the syndrome described here. As a consequence, we raise the hypothesis of a gain-of-function effect of this variant.
In conclusion, de novo mutations in exons 3 and 4 of the X-linked gene TFE3 are responsible for a neurocutaneous disorder with specific and recognisable facial dysmorphism, lysosomal storage disorder-like features and PM. This series unravels TFE3 as a major gene responsible for HMI and for a rare cause of syndromic ID. Furthermore, we provide clinical and molecular data on a previously unidentified lysosomal storage disorder, in which new insights, especially biochemical features, will probably be investigated further, together with the description of more patients. Further delineation of this phenotype will indeed allow a better understanding of the link between lysosomal signalling and development. Finally, the evidence for mosaicism in this recently described disorder highlights the importance of considering mosaic variants on next-generation sequencing reports in diagnostic, including for patients without suggestive phenotype.
Acknowledgments
The authors wish to thank the subjects and families involved in the study and the University of Burgundy Centre de Calcul (CCuB, see Web Resources) for technical support and management of the informatics platform. Several authors of this publication are members of the European Reference Network for Developmental Anomalies and Intellectual Disability.
References
Footnotes
Contributors DL wrote the paper. AS, MA, VC, MC, ET, BC, IC, LD, YD, OE, MJGS, TJ, PK, JS-O, JT, KvG, EV, BM, ToBa and RAJ performed and interpreted the molecular data. TaBi, JC-S, EDB, FD, AF, DG, AG, DJH, MH, BI, KL, VLR, LP, KS-S, IV, MMvH, SvK, CWH, YA, EK, MM, SM, YM, HP, LS and PZ referred the patients. PV, JB, CT-R, JB-R and LF planned and supervised the work.
Funding This work was funded by the Agence Nationale de la Recherche (ANR-13-PDOC-0029 to J-BR), the Programme Hospitalier de Recherche Clinique National 2010 (NCT01950975 to PV), the Conseil Régional de Bourgogne through the Plan d'actions Régional pour l'innovation and the European Union through the PO FEDER-FSE Bourgogne 2014/2020 programmes to the GAD team. This study was supported by the URDCat programme (PERIS SLT002/16/00174). EV was funded by a grant from the Ministerio de Economia, Industria y Competividad (Juan de la Cierva programme FJCI-2016-28811).
Competing interests MJGS and ET are employees of GeneDx, Inc.
Patient consent for publication Parental/guardian consent obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository.