Article Text

Abstract
Background Subtelomeres are variable regions between telomeres and chromosomal-specific regions. One of the most studied pathologies linked to subtelomeric imbalance is facioscapulohumeral dystrophy (FSHD). In most cases, this disease involves shortening of an array of D4Z4 macrosatellite elements at the 4q35 locus. The disease also segregates with a specific A-type haplotype containing a degenerated polyadenylation signal distal to the last repeat followed by a repetitive array of β-satellite elements. This classification applies to most patients with FSHD. A subset of patients called FSHD2 escapes this definition and carries a mutation in the SMCHD1 gene. We also recently described patients carrying a complex rearrangement consisting of a cis-duplication of the distal 4q35 locus identified by molecular combing.
Methods Using this high-resolution technology, we further investigated the organisation of the 4q35 region linked to the disease and the 10q26 locus presenting with 98% of homology in controls and patients.
Results Our analyses reveal a broad variability in size of the different elements composing these loci highlighting the complexity of these subtelomeres and the difficulty for genomic assembly. Out of the 1029 DNA samples analysed in our centre in the last 7 years, we also identified 54 cases clinically diagnosed with FSHD carrying complex genotypes. This includes mosaic patients, patients with deletions of the proximal 4q region and 23 cases with an atypical chromosome 10 pattern, infrequently found in the control population and never reported before.
Conclusion Overall, this work underlines the complexity of these loci challenging the diagnosis and genetic counselling for this disease.
- facio scapulo humeral dystrophy
- molecular combing
- smchd1
- subtelomeres
Statistics from Altmetric.com
Introduction
Subtelomeres are highly variable DNA sequences lying at the interface between telomeres and chromosome-specific regions. The rate of recombination at subtelomeres is higher than in the rest of the genome and subtelomeric variation contributes to genome variability and to a wide range of diseases from acquired and common ones to rare genetically inherited syndromes.1–3 In addition, these telomere-adjacent DNA sequences are crucial for telomere regulation and integrity.4 Subtelomeres contain both coding and non-coding transcripts and promoters for the non-coding telomeric repeat-containing RNA transcribed from the subtelomere into the (TTAGGG)n telomeric tract.5 Composition of subtelomeric regions in terms of sequence (haplotype) and DNA copy number contribute to the higher-order chromatin organisation of these chromosomal regions, abutting telomeres, and regulation of genes in close proximity by telomere position effect6–9 or at a long distance, in a discontinuous manner through formation of long-distance loops.10–13
Facioscapulohumeral dystrophy (FSHD) is one of the most puzzling genetic diseases linked to subtelomeric imbalance. In the majority of patients (FSHD1, 95%), this autosomal dominant neuromuscular disorder ranked as the most common neuromuscular hereditary disorder with a prevalence of 1:8000–1:20 000 is not linked to a mutation affecting the coding sequence of a protein involved in muscle function but to a deletion of an integral number of repetitive D4Z4 macrosatellites at the 4q35 chromosome end.14 15 Clinically, symptoms usually arise between the age of 20–40 years with a typical asymmetrical weakness of facial, scapular girdle, upper limb and lower extremities muscles.16
The locus linked to the pathology is located in the subtelomeric region of the 4q arm.17 18 In the control population, the number of repeated units of this 3.3 kb GC-rich element is between 11 and 150 copies (eg, >35 kb and up to an estimated size of 495 kb) whereas in patients with FSHD1, one of the D4Z4 arrays carried by the 4q35 allele is contracted and contains between 1 and 10 units with a threshold size <35 kb.19 Distal to D4Z4, two main sequences have been described, termed qA and qB haplotypes.20 The 4qA sequence is characterised by the presence of a 260 bp sequence called pLAM containing a degenerated polyadenylation signal for the DUX4 retrogene encoded by D4Z4. The pLAM is followed by an estimated array of 6.2 kb containing tandem copies of 68 bp ß-satellites.20–22 The 4qB sequence is 92% homologous to 4qA but contains a LINE sequence. The 10q26 region is approximately 98% identical to the 4qA end in the 40 kb proximal to D4Z4 and at least 10 kb distal to the repeat.21 23 24 On this chromosome, the number of D4Z4 is variable. In addition, SSLP analyses of the 4qter region revealed the existence of microsatellites of different sizes upstream of D4Z4 with at least three different haplotypes for the 4qA allele and six haplotypes for 4qB.24 The current model explaining the pathogenesis of the disease postulates that in FSHD1, chromatin relaxation linked to shortening of the D4Z4 array or to mutation in SMCHD1 in FSHD2 causes overexpression of the DUX4 transcript encoded by D4Z4. DUX4 is transcribed from the last D4Z4 repeat and through the distal pLAM sequence containing a degenerated polyadenylation site required for stabilisation of the transcript and production of the DUX4 protein.
In most routine laboratories, FSHD diagnosis is based on a Southern blot (SB) technique after digestion of genomic DNA with the EcoRI enzyme and hybridisation of the p13E-11 probe (D4F104S1) that maps to the proximal region adjacent to the first D4Z4 repeat. However, in approximately 20% of cases, this technique fails to provide a clear conclusion regarding the number of repeated units and haplotype and remains inconclusive due, for instance, to somatic mosaicism, 4q-10q translocations, p13E-11 deletion or existence of other non-canonical variants.25 26 To bypass these limitations and provide a method allowing a direct assessment of the size of the D4Z4 array on both the 4q and 10q chromosomes together with the type of haplotype, we have developed a molecular combing (MC)-based strategy.27 This technology originally developed to map genes for positional cloning and widely used to study DNA replication shares most of the advantages of FISH, with a 100-fold improved resolution.28 In addition, MC allows the direct visualisation and cartography of numerous individual DNA molecules at a resolution of 1 kb28 29 and a high reproducibility due to the constant stretching of DNA molecules on glass slides. For FSHD, its main advantage is to allow the direct visualisation of the relevant 4q35 and 10q26 loci27 and appeared as a powerful tool for molecular diagnosis of FSHD and resolution of complex cases.25 27
Here, we exploited data gathered from hundreds of individuals analysed by DNA combing to determine the genetic organisation of the 4q35 and 10q26 loci. By analysing more than 400 4q and 10q alleles, we determined the mean size of D4Z4 arrays in the different contexts, the size and distribution of the qA-specific and qB-specific sequences and organisation of the region upstream of the D4Z4 repeats. Our results reveal an important variability between samples and the complexity in realising a complete assembly of these subtelomeric regions. Subsequently, we report analyses of individuals clinically affected with FSHD and displaying atypical genotypes, such as 4q mosaicism or p13E-11 probe deletion. We also report 23 cases clinically diagnosed with FSHD and carrying an atypical chromosome 10 pattern, infrequently found in the control population and never reported before.
Statement of objectives
Exploit the resolution provided by MC and bar coding of the 4q35 and 10q26 subtelomeric loci to uncover the complexity of these regions in individuals affected with FSHD and in the general population.
Materials and methods
Materials and Methods are detailed in the online supplementary information section.
Supplemental material
Results and discussion
Analysis strategy
Since the validation of MC for the molecular diagnosis of FSHD,27 we have processed 1029 blood samples from index cases or relatives (figure 1A–D). A number of cases were referred to our Medical Genetics Department for FSHD molecular diagnosis and exclusion diagnosis or familial segregation studies. After initial steps of validation of the technique,27 we have used the combing methodology to test in priority patients for whom ambiguous results were obtained by SB (either positive or negative). The major advantage of MC is the direct visualisation of the haplotypes allowing a straightforward interpretation, especially in complex situations. For SB, we considered unequivocal blotting results where four alleles (two signals for 4q and 10q alleles) were clearly visible and additional alleles were absent. Atypical profiles systematically led to the processing of the blood sample by MC. We considered as atypical profiles the absence of one of the four alleles, suggestive of a proximal deletion, the presence of an additional band suggesting the presence of an additional allele or mosaicism and samples for which we observed a discrepancy between the molecular diagnosis results and those expected to carry the genomic anomaly according to the clinical data but showing absence of the short D4Z4 repeat. We also included patients carrying a contracted 4q allele with a size close to the pathological threshold (greater or equal to eight units) in order to verify the association of this shortened D4Z4 array with the distal qA variant. In addition, since the development of the test, a proportion of patients were tested directly by MC. Those correspond, for example, to relatives of an index case previously explored with an unequivocal result when segregation of the pathological allele was needed. A number of cases were also diagnosed directly by MC.

Schematic representation of the 4q35 and 10q36 loci and respective bar codes. (A) Schematic representation of the 4q35 subtelomeric locus. from left to right, the FRG1, TUBB4Q and FRG2 genes are indicated. Sequences starting with an inverted D4Z4 repeat (green arrow), are specific to the 4q35 locus (red lines) while regions located between the D4Z4 array and the inverted D4Z4 repeat are also present on chromosome 10 (10q26 locus). The D4Z4 array is depicted by green triangles. The 4qA and 4qB haplotypes correspond to different genomic elements distal to D4Z4. The 4qA (red rectangle) is characterised by the presence of a sequence named pLAM immediately distal to the last D4Z4 repeat and followed by an array of repeated β-satellite elements associated with a 4qA haplotype upstream of the telomere (red arrows). The 4qB allele (depicted as a blue rectangle) differs from the 4qA by the absence of β-satellite elements upstream of the telomere (red arrows). (B) Illustration of the V3 pink bar code used to distinguish the two 4q alleles (qA/B) based on a combination of four different colours and different DNA probes encompassing the distal regions up to the telomeric sequence as previously described.27 This four-colour bar code comprises one probe detected in blue and one in pink, which hybridise the proximal region common to chromosomes 4 and 10, one 6 kb probe detected in red, which hybridises the (TTAGGG)n telomeric ends and a red probe that hybridises the qA-specific β-satellite region. The qB-specific probe, immediately adjacent to D4Z4, is detected in blue. The proximal 4q-specific region is detected by a combination of red and pink probes. (C) Schematic representation of the 10q36 locus. This locus shares 98% of homology with the 4q35 locus (dashed arrow) starting from a truncated inverted D4Z4 repeat (green arrow) and the same organisation in its distal part, with a variable-length D4Z4 array A-type and B-type haplotype abutting the telomere. (D) Illustration of the V3 pink bar code used to distinguish the two 10q alleles (qA/B) based on a combination of four different colours and different DNA probes encompassing the distal regions up to the telomeric sequence. The bar codes for the 4q-10q homologous regions are identical. The proximal 10q-specific region is identified by hybridisation with a blue probe. (E) Out of the 1029 patients analysed, 92.5% showed a normal profile with four distinct alleles and absence (61%, 627 cases) or presence (31.6%, 318 cases) of D4Z4 array contraction on a 4qA allele. We identified 7.7% of cases with an atypical profile with 2.7% of patients with a mosaic D4Z4 array contraction, 2.14% of patients with a 4qA cis-duplication, 0.7% of cases with a deletion of the p13E-11 probe and 1.5% with either a surnumerary 10q allele, a complex rearrangement of the 10q chromosome or both a 4q and 10q rearrangement. (F) We plotted the number of residual D4Z4 repeats of the shortest 4q35 allele in patients with FSHD2 carrying a mutation in SMCHD1 (grey circles), in patients carrying a cis-duplication of the 4q35 region from patients described in25 and newly diagnosed patients (nine cases) (red triangles, patients with white filling are carrier of a SMCHD1 mutation) and patients in which we found an additional copy of chromosome 10 (blue diamonds, table 1).
The diagnosis of FSHD1 was confirmed in 32.26% of the samples tested for whom only one contracted 4qA allele was unambiguously present (332 cases out of the 1029 cases analysed, table 1). Presence of a short D4Z4 allele or FSHD1 was discarded for 61% of the individuals tested. In this group, the four alleles (4q and 10q) were unambiguously distinguished, with no contracted 4qA allele or other variant. This second group includes individuals clinically affected with FSHD classified as FSHD2 (figure 1F). In this subgroup of 627 samples, we identified 15 cases carrying a mutation in SMCHD1 (1.45% of the total cohort of 1029 samples and 3.5% of affected cases). Cases referred to our centre for exclusion diagnosis are individuals presenting with an undiagnosed neuromuscular disorder. Other cases are individuals explored in case of familial segregation analysis or prenatal testing.
Summary of molecular combing (MC) data for analysis of 1029 cases comprising 426 individuals diagnosed with FSHD.
Among the 1029 patients reported here, we previously described complex rearrangements consisting of a cis-duplication of a long D4Z4 array (>35 kb in most cases) and a distal short D4Z4 array (<35 kb) in 14 patients affected with FSHD.25 Nine additional patients have been diagnosed with the same type of cis-duplication. This group of patients represents 2.14% of the 1029 cases analysed and 5.38% of all patients described in this cohort. Besides, we found a number of additional atypical genotypes including mosaic cases (2.6% of the total number of cases; 6.34% of patients with FSHD), deletion of the p13E-11 probe (0.7% of the total number of cases; 1.64% of patients with FSHD). We also found a significant number of patients harbouring the presence of an additional 10q allele (1.5% of the total number of cases; 3.75% of patients with FSHD) (table 1).
Determination of the D4Z4 array size by MC at the 4q and 10q subtelomeres
Estimation of the number of D4Z4 repeats at the 4q and 10q loci have been mainly based on SB analyses, with a low resolution especially for large DNA fragments. MC facilitates high-resolution analysis of a given genomic region thanks to the combination of specific DNA probes and the constant stretching of DNA molecules on glass slides.28 29 Moreover, standardisation of the processing and analysis facilitates in-depth characterisation of complex genomic regions. We thus took advantage of this methodology to determine the size of the long and short D4Z4 array on 4qA and 4qB chromosomes (figure 2A). For A-type haplotypes, the mean size of long D4Z4 arrays (>35 kb) is 108.9 kb (33 units) and ranges between 35 kb and 338 kb (11 to 102 units) (online supplementary table 1). In patients with FSHD1, the mean size of the short D4Z4 allele is 18 kb corresponding to five units.
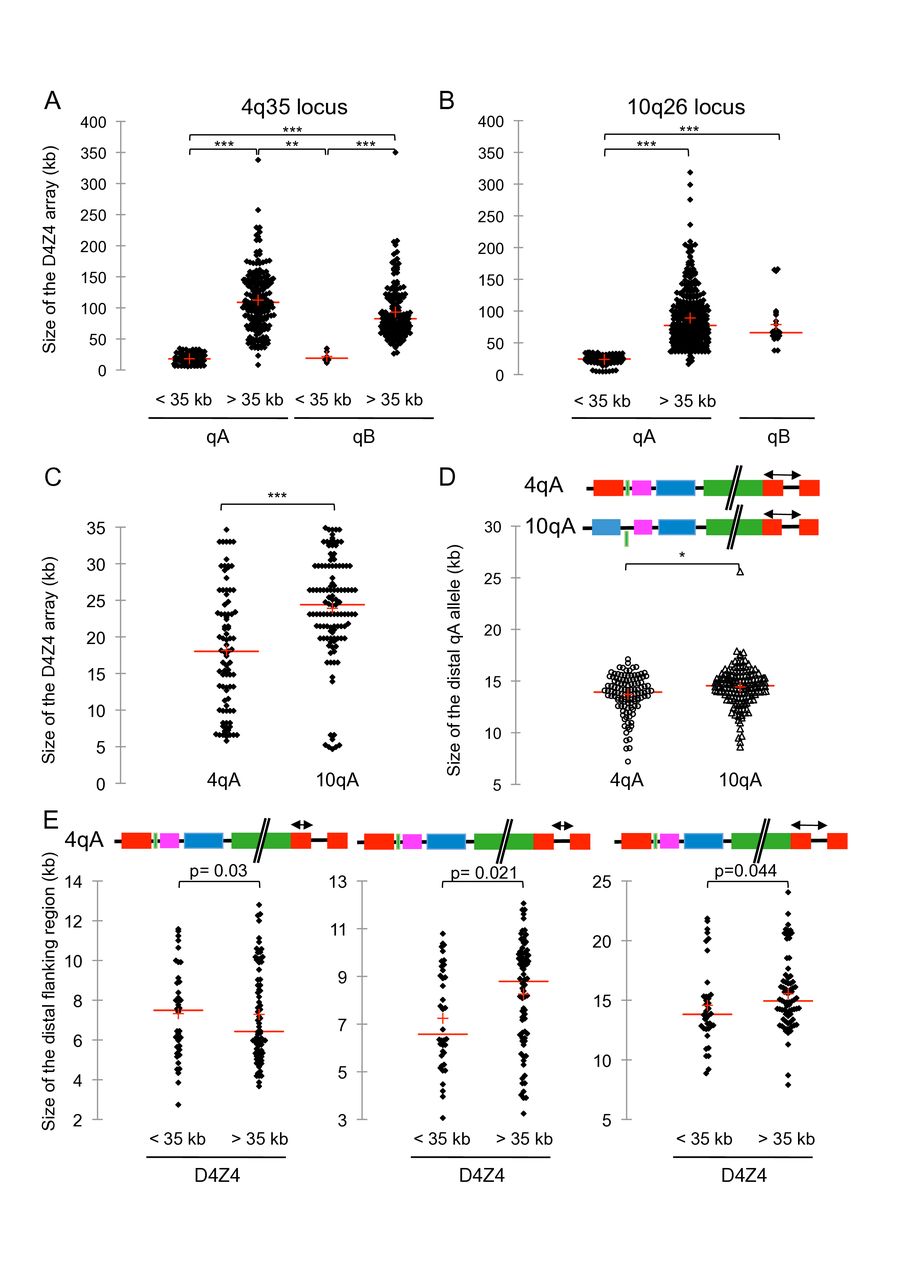
Sequence length variation at the distal 4q and 10q subtelomeres. (A) We determined the distribution of the D4Z4 array size carried by chromosome 4 by analysing signals obtained by molecular combing (MC) in 218 individuals either affected or non-affected with facioscapulohumeral dystrophy (FSHD). In all cases the two 4q and two 10q alleles were analysed independently, that is, 436 alleles for each chromosome. Scattergrams display the size distribution. The red line corresponds to the mean size in the different subgroups: short D4Z4 arrays (<35 kb on A-type chromosomes, n=86; mean size=18.025 kb); long D4Z4 arrays (>35 kb on A-type chromosomes, n=193; mean size=108.9 kb); short D4Z4 arrays (<35 kb on B-type chromosomes, n=8; mean size=23.925 kb); long D4Z4 arrays (>35 kb on B-type chromosomes, n=149; mean size=83 kb). (B) Scattergrams of D4Z4 array size carried by chromosome 10 in 218 individuals for short D4Z4 arrays (<35 kb on A-type chromosomes, n=114; mean size=24.4 kb); long D4Z4 arrays (<35 kb on A-type chromosomes, n=299; mean size=79.2 kb) and B-type chromosomes, (n=25; mean size=66 kb). (C) Size comparison between short D4Z4 arrays (<35 kb) on 4qA (n=86; mean size=18.025 kb; five D4Z4 units on average) and 10qA (n=114; mean size=24.4 kb; seven D4Z4 units, on average). (D–E) Schematic representation of the 4q and 10q chromosomes with the region analysed indicated by an arrow. supplementary tables 3–6. The size is indicated in kilobases (kb). The mean size is shown by the red line. Differences in size distribution were determined using a non-parametrical Kruskal-Wallis test with pairwise comparisons and Bonferroni correction for false positives. ***, p<0.0001; **, p=0.002, *, p=0.003. (D) We analysed the region distal to D4Z4 containing β-satellite elements distal to 4qA-type (n=114) or 10qA-type (n=158) chromosomes. (E) Scattergrams of the qA distal region containing β-satellite element (left graph), distal gap (middle graph) and total qA region on chromosome 4 with short (<35 kb) or long (>35 kb) D4Z4 arrays.
We also analysed a large number of 4qB alleles with size ranging between 3 and 106 repeated D4Z4 units (11.6–350 kb). Interestingly, we only observed a low number of short 4qB alleles (eight alleles) and a broader size dispersion for 4qA alleles compared with 4qB, with a significantly smaller size for D4Z4 arrays on 4qA-type alleles compared with B-type alleles (p=0.002; figure 2A).
For the 10q region, the median size of the D4Z4 array is 24 kb for the 10qA short array (≤35 kb; 7 D4Z4 units, n=109), 89 kb for the 10qA long array (>35 kb; 27 D4Z4 units, n=304), and as observed for 4q alleles, significantly smaller for the 10qB long array (>35 kb; 24 D4Z4 units, n=25; p<0.001), (figure 2B; online supplementary table 2).
By comparing D4Z4 arrays on the 4qA versus 10qA chromosomes, we observed a significant difference in size (p<0.001) (figure 2C). D4Z4 long arrays are usually in a range comprised between 11 and 100 repeats and rarely reaches 150 units as suggested in the literature. Interestingly, the distribution of 10q-type arrays is different. D4Z4 arrays on the 10q chromosome are smaller compared with 4q with a vast majority of alleles comprised between 11 and 42 units (>35 kb to 140 kb).
Identification of 10qB alleles
Furthermore, we have identified a total of 25 alleles from chromosome 10q carrying a B-type variant and representing 5.7% of the 438 chromosomes 10 analysed. In this category, the size of the D4Z4 array ranges between 38 kb and 166 kb (11–50 units), in the same size range as the 10qA alleles. We did not detect any short 10qB allele.
So far, the proportion of 10qB alleles has been underestimated since no attempt to determine their frequency in large cohorts has been made. Given the common origin between 4q and 10q alleles, 10qB alleles likely result from the translocation of 4qB alleles as hypothesised for 4qA-10qA translocations. In a previous study of a large cohort of subjects in the general population, 10qB frequency was estimated to be 5%, a percentage slightly lower than the percentage that we have estimated by MC (5.7%). This slight underestimation might be explained by the assumption that 10q chromosomes were exclusively of type A20 24 and by absence of systematic testing of 10qA-type or B-type alleles in SB for FSHD diagnostic purposes.23 Indeed, interpretation of HindIII-qA/qB blots is often complex since it is based on the comparative size analysis of four different alleles after hybridisation with the p13E-11 probe and determination of the EcoRI and HindIII fragments, which are not linear. In addition, as detailed below (figure 2D–E), the HindIII-qA-type fragment is highly polymorphic due to the repetitive nature of the 68 bp β-satellite region while the HindIII-qB fragment devoid of these short tandem repeats is less polymorphic in size.
MC for exploration of the 4q and 10q distal subtelomeric regions
Despite the global assembly of the human genome, subtelomeric regions remain partially sequenced due to segmental duplications and variability in haplotypes.1–3 At 4q and 10q, the distance between the D4Z4 repeat and the telomere has been estimated between 25 kb and 40 kb but the sequences downstream of D4Z4 are poorly described and only partially sequenced.1 3 We took advantage of MC to explore these regions at 4q and 10q and determine the size between the end of the D4Z4 array and the telomere (online supplementary tables 3–6). More precisely, we measured the size of the type A allele comprising the ß satellite array (figure 2D–E; online supplementary tables 3;4), the size of the B-type allele (online supplementary tables 5;6), the size of the telomeric signal and gaps between the different probes for the 4q and 10q regions (online supplementary tables 3–6). Regardless of the number of D4Z4 repeats, the size of the ß satellite-containing region and abutting gap is significantly smaller at the 4q locus compared with the 10q (p=0.05) (figure 2D).
By comparing the 4qA distal region between short and long D4Z4 alleles, we observed significant size differences. The median size for the ß satellite-containing region is larger than the previous estimations of 6.2 kb with a mean size of 7.5 kb. The size of the subtelomeric sequence between the end of the D4Z4 array and telomere is larger for the short 4qA D4Z4 arrays compared with the long 4qA arrays (22.5 kb vs 17.5 kb, respectively, figure 2E) with the distance between the last D4Z4 repeat and the 4qter telomere smaller than the previous estimation of 25–40 kb. The complexity of this subtelomeric region also reveals an increased distance between the last D4Z4 repeat and the telomere in patients with FSHD compared with healthy individuals.
The median size of ß satellite-containing sequence is 5.33 kb and 6.12 kb respectively for short 10qA D4Z4 array (≤35 kb; n=38) and long 10qA array (>35 kb; n=119) with a significant difference between the two groups (p-values 0.015) (online supplementary tables 4;5). The median size of ß satellite-containing sequence and the upstream gap is 13.82 kb for short 10qA D4Z4 array (≤35 kb; n=38) and 14.63 kb for 10qA long array (>35 kb; n=123) with a significant difference between the two groups (p=0.019). The median length of the β-satellite sequence, the gap and the telomeric sequence is significantly different between the short 10qA D4Z4 array (≤35 kb; n=38; 23.9 kb) and the long 10qA array (>35 kb; n=123; 25.02 kb, p=0.017) (online supplementary table 4).
Characterisation of patients with FSHD with complex genotypes
Between 2012 and 2017, we analysed 1029 individuals by MC, either for FSHD molecular diagnosis, exclusion diagnosis or familial segregation studies. We identified atypical 4q or 10q genotypes in 8.7% of the cases (figure 1). This subcategory includes individuals carrying a cis-duplication of the 4q35 region that we initially described in 15 individuals corresponding to 14 patients affected with FSHD and 1 non-affected carrier.25 Nine additional cases have been characterised with the same cis-duplication since our previous publication.25 Among the rest of these 8.7% of cases, somatic mosaicism was detected in 2.8% of patients presenting with clinical signs of FSHD (online supplementary table 7). For each patient, the percentage of mosaicism was determined by counting the proportion of short versus long D4Z4 arrays. This percentage ranges from 6% to 52% of cells carrying a short D4Z4 allele. All patients displayed a significant decrease in D4Z4 methylation (Roche et al, submitted) and none of the 12 patients analysed by whole-exome sequencing were carriers of an SMCHD1 pathogenic mutation suggesting that the short 4qA allele is pathogenic. Of note, two of these patients presented with 4q mosaicism segregating with a complex 10qA allele consisting of a cis-duplication of a long D4Z4 array (80 kb, 24 D4Z4 units) followed by a sequence corresponding to the A-type probe and a second array of 3 kb (1 D4Z4 unit) followed by an A-type probe.
Characterisation of the 4q region and identification of patients with deletion of the p13E-11 probe region
We have identified four cases of large deletions encompassing part of the D4Z4 array and the proximal region that hybridises the p13E-11 probe (figure 3, online supplementary table 8).
The first index case (patient 100 519A) is a patient of Lebanese origin affected with a typical familial form of FSHD. The index case carries a D4Z4 array of 18 kb (five units) that segregates with deletion of the p13E-11 probe region on the same chromosome. The patient’s relatives have been also been explored by MC. The deleted allele is present in three out of six siblings who are also affected with FSHD. The deletion was transmitted from the father also affected by the disease (figure 3A). The second case (1211 108A, online supplementary table 8) carries a proximal deletion of the p13E-11 probe region together with a short three unit D4Z4 array.

Proximal deletions of the p13E-11 and D4Z4 regions. (A) Pedigree of the family 100 519A with four members in the third generation and one member in the second generation. Representative images of the molecular combing (MC) analysis of patient 00519A-III1 (index case) affected with facioscapulohumeral dystrophy (FSHD). The patient carries a short D4Z4 array and a large deletion of the proximal region encompassing the blue and pink probes. (B) Presentation of other patients identified with a deletion of the p13E-11 probe. The deleted 4q allele is visualised by the lack of hybridisation of the blue probe corresponding to the p13E-11 region proximal to D4Z4 and the presence of the proximal red probe specific for the 4q chromosome and the downstream green probe corresponding to D4Z4. In the two patients presented here, the deletion of the proximal region is of different size, with a deletion of the blue probe (patient 14668) or a deletion of the blue and pink probes (patient 19185). Of note, these two patients carry a >11 D4Z4 repeats array. In patient 14668, the second 4q allele (4qA) contains 21 repeated units. In patient 19185, the second allele (4qA) contains 51 repeated units. (C) We describe here a complex situation with a patient presenting with an insertion of an 8 kb D4Z4 repeat (two units) upstream of the pink probe on a type B 4q allele with 24 repeated units. The second 4q allele contains 35 D4Z4 units.
In these two cases, the proximal deletion segregates with a short D4Z4 which is likely pathogenic. DNA methylation was tested in these two patients for the DR1 region by sodium bisulfite sequencing and levels were comparable to those observed in patients with FSHD1 (50% and 44.1% of methylated CG, respectively).
The third (14 668) and fourth (19 185) patients are affected with FSHD and carry a deletion of the p13E-11 probe region that segregates with a 12 D4Z4 repetitive array (figure 3B, online supplementary table 8) and thus do not correspond to the definition of FSHD1. Patient 14 668 displays a decreased methylation (36% for the DR1 region) while no hypomethylation was found in the other case (19185; 63.9% of methylated CG). We excluded FSHD2 in these patients by absence of mutation in SMCHD1 assessed by whole-exome sequencing (online supplementary table 8) indicating that the deletion of the proximal region is associated with the disease despite the absence of short 4qA allele.
In patients 100 519A and 19 185, the deletion also encompasses the magenta probe of 10 kb proximal to the first D4Z4 repeat with the red probe abutting directly the first macrosatellite. In these cases, and based on the size of the probes, the size of the deleted region upstream of the first D4Z4 repeat is estimated as least of 35 kb.
In this subcategory of patients carrying proximal 4q deletions, we also report a complex case carrying a long (25 units) D4Z4 repeat on the 4q chromosome and a complex rearrangement of the other 4q chromosome consisting of a cis-duplication of a B-type allele with a duplication of one D4Z4 unit followed by a B-type sequence in the proximal part of the repeat followed by a 37 units repeat followed by a B-type sequence (figure 3C). By the absence of a pathogenic variant in SMCHD1, we excluded FSHD2 in this patient (online supplementary table 9).
Based on SB analysis and the presence of a single allele after hybridisation with the p13E-11 probe, the frequency of non-canonical deletions that extend into the D4F104S1 hybridisation region has been estimated to be 3% of FSHD cases.30 Deletions of up to 78 kb upstream of D4Z4 have been reported31 with phenotypes compatible with the FSHD clinical spectrum. In case of p13E-11 deletions, SB in PFGE is more efficient to visualise the absence of an allele and detection of the other alleles but remains unsatisfactory and a source of underdiagnosis which might require additional SB experiments and the use of other probes like the 9B6A probe that hybridises to D4Z4.32
MC is thus an interesting tool since the four alleles are directly visible and deletion easily identified by absence of the proximal blue probe common to both 10q and 4q alleles and the presence upstream of this probe of the 4q-specific (red) or 10q-specific (blue) probes to exclude breakage of the DNA fibre. Thus, the frequency and size of proximal deletions may be better estimated by MC. In our cohort of patients, we have identified four patients with deletion of this proximal region and one patient with a complex rearrangement. Of note, in the five patients reported here, we have not found any mutation in SMCHD1, including in patients carrying a D4Z4 array >11 units indicating that deletion of the proximal region associated or not with a short D4Z4 array segregates with typical signs of the disease. The frequent occurrence of 4q proximal deletions highlights the importance of this region in the regulation of the locus with the possible presence of regulatory elements such as those corresponding to permissive haplotypes24 or putative regulatory elements33 34 which remain to be fully characterised.
Analysis of the 4q and 10q proximal regions
Through our analyses of the 4q and 10q chromosome ends, we noticed a large variability in the regions hybridised by the proximal probes. We thus analysed the size of the different probes and gaps corresponding to the D4F104S1 (p13E-11) and 4q-specific or 10q-specific regions upstream of the first D4Z4 (online supplementary tables 10;11, online supplementary figure 3). The size of the gap upstream of the first D4Z4 repeat is identical in the different groups of alleles (online supplementary figure 3A, online supplementary table 11). The p13E-11 probe (blue) with an estimated size of 20 kb is in the same size range for long, short A-type or B-type 4q alleles (11–37.7 kb) but is more variable for the different 10q chromosomes, ranging between 24.8 kb to 62.9 kb. An important variability is also observed for the gap located between the blue and magenta probes with an estimated size of 5 kb, at the 4q end with a size ranging from 3.7 kb to 26.6 kb, compared with 4.6 kb to 15 kb for the 10q chromosome (online supplementary figure 3C, online supplementary table 11). By individual analysis of the different regions, we did not observe any significant difference in size in the proximal part of the 4q region between the different types of alleles. However, by combining analyses of the different probes and gaps, between the short 4qA and 10qA alleles, we observed a significant difference in size indicating that despite the estimated 98% of homology between these subtelomeric regions, the proximal region is also highly variable (figure 4, p=0.043). Altogether these analyses suggest that despite a high conservation between the two chromosome ends, likely due to a 4q-10q duplication during evolution, these subtelomeres are highly prone to recombination and evolved independently.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sequence length variation at the proximal 4q and 10q subtelomeres. (A) Schematic representation of the molecular combing bar code with position of the region. As indicated by an arrow, we analysed the length of the centromeric portion of the 4q35 and 10q36 loci. (B) Scattergrams display the size distribution for the different types of alleles. The red line corresponds to the mean size. The proximal region is identical for the different types of 4q alleles but more variable between 10q alleles as indicated by the distribution. The proximal region is significantly longer for short 4qA alleles compared with short 10qA.
Frequent detection of triple 10q signals in patients with FSHD
Unexpectedly, we have identified additional alleles in a number of patients for the bar code corresponding to the 10q allele (table 2). These additional chromosome 10 alleles have not been reported previously and might have been unnoticed so far by classical SB analyses. All contained a repetitive D4Z4 array associated with a distal variant qA region. The observed 10q signals are always independent of the two other 10q alleles that segregate in a Mendelian way for all samples. In addition, the number of 10q signals corresponding to this additional allele is high in all cases indicating the absence of artefact and suggesting that these alleles are equally represented.
Individuals carrying three copies of the 10q26 locus. We tested the presence of the short or long pLAM sequence associated with a 4qA sequence in most of the cases presented here (underlined). Data are presented in online supplementary figure 5.
One hypothesis is that these 10q alleles correspond to somatic mosaics, as seen for chromosome 4. Indeed, the frequency of chromosome 4 mitotic rearrangements is high and given the homology between the 10q and 4qter regions and occurrence of somatic rearrangement between these two regions,35 somatic D4Z4 contraction might likely occur on chromosome 10 as well.
However, in at least three of the families investigated (families 3, 4 and 5, table 2), these 10q alleles are transmitted from the first to the second generation and co-segregate with a normal parental 10q allele, excluding mosaicism in these cases. In family 4, the father and the son carry a seven-unit 4qA contracted allele and are affected with FSHD. In addition, the father carries three 10q alleles of which a 10qA allele has one to two D4Z4 units. This allele was transmitted to his two children independently of the FSHD allele arguing in this family against the implication of this supernumerary allele in FSHD and favouring the hypothesis of the presence of a genomic variant with a 10q26 duplication, not visible by MC. This additional allele could correspond to a complex rearrangement of the 10qter with a cis-duplication of the region containing D4Z4, the proximal chromosome 10-specific sequences, and the distal qA sequence as described for chromosome 4.25
In family 3, the supernumerary 10q allele (10qA at 2 units) segregates with the same maternal 10q allele (10qA at 29 units) in both sons. In this case again we cannot exclude the presence of a cis-duplication.
In the remaining kindreds, the size of stretched DNA molecules and the absence of detection of large size 10qter signal as described for the 4qter region25 is not in favour of tandem duplications. To retain the explanation of a cis-duplication, it would be necessary to imagine that the two 10q signals are at a distance of 300 kb or more and thus always separated by a random break occurring between the two during the stretching process and therefore never visible on the same fibre. This hypothesis is not likely given the number of signals analysed in the different cases presented. Another alternative would be that the additional 10q allele is located on another chromosome, by duplication of the region and insertion into another region of the genome. We tested this hypothesis by performing FISH, an experiment on metaphase chromosomes, a technique suitable for the analysis of chromosomal rearrangements of large sizes. The D4Z4 probe hybridises to chromosomes 4 and 10 with a variable intensity likely dependent on the size of the D4Z4 array. We did not observe any additional spot on another chromosome excluding the presence of an additional contracted 10q allele. If the two loci were duplicated in tandem and are at least 3 Mb apart, the resolution of FISH would allow seeing two distinct spots on the same chromosome. In this scheme, the two loci would be too distant to be seen together by MC and too close to be distinguished by two spots by FISH. If the two loci were duplicated in tandem and close enough to give only one signal in FISH, the difference in signal intensity between the two alleles would not be visible in FISH but likely visible by MC.
Contractions of D4Z4 on chromosome 10q are not a priori pathogenic and accordingly, in a number of cases presented here, the additional 10q contracted allele does not always segregate with FSHD, so its involvement in FSHD pathogenesis is questionable. Nevertheless in six cases with clinical FSHD, the additional short 10qA allele segregates with the disease and two long 4qA alleles suggesting that it might be causative. None of these six patients carries a mutation in the SMCHD1 gene. Of note, in this subgroup of affected individuals, several cases carry a short 4qB-type allele together with a long (>20 units) 4qA-type allele (case 15 573 [family 5-II1], B2014-2538, B2015-0851, B2016-1344) (table 2). One of these patients, also carries a cis-duplication of the 4qter region with a stretch of 20 D4Z4 elements followed by a nine repeats array on a type A chromosome. Interestingly, these individuals developed FSHD during childhood suggesting that this short 10q allele might contribute to the disease or act as a modifier in disease severity.
Detection of the permissive pLAM sequence in patients with atypical genotypes
In our cohort of atypical cases, we tested the presence of the pLAM sequence associated with stabilisation of the DUX4 transcript produced from the last D4Z4 unit and adjacent A-type sequence. Primers for the short 4qAS or long 4qAL sequences, specific to A-type alleles and unable to amplify 4qB or 10qA alleles have been previously described.36 We were able to amplify the 4qAS fragment in 19 out of the 20 atypical cases tested and detected only the 4qAL variant in the remaining sample (sample 4, online supplementary figure 5). For five individuals, both the 4qAS and 4qAL fragments were detected (samples 2, 16, 19, 21, 22). All PCR fragments were sequenced and match the previously reported sequences.36
Concluding remarks
Using MC, we explored a large cohort of patients affected with FSHD for whom the genetic cause of the disease had not been resolved and estimated to be approximately 20% of clinically affected individuals. Among those, we previously reported the existence of cis-duplications of the 4qter region present in 2%–2.9% of the population25 37 and in 15 individuals corresponding to 10 FSHD families. The duplication segregates with a short D4Z4 array on the second 4q allele in 3/11 cases or mutation in SMCHD1 in 5/10 families suggesting that it might be associated with FSHD1 or FSHD2, respectively. Yet, the cis-duplication is the only genetic defect that segregates with clinical signs of the disease in 2/10 of our cases and in a proportion of cases later reported by Lemmers et al 37 suggesting that it might also be causal in a small number of patients.25 Since our initial publication describing this recurrent 4q35 rearrangement, we have identified nine additional patients displaying the same cis-duplication of the 4q35 region, accounting for 5.16% of all clinically affected cases reported in this study.
In addition, we report here the presence in patients clinically affected with FSHD of typical 4q alleles with deletion of the region proximal to the first D4Z4 repeat (D4F104S1), including in patients carrying a normal D4Z4 array size (three out of six). We also report the presence of an additional 10q allele in 11 unrelated families. Based on our MC data, cis-duplications are less frequent on chromosome 10 than on chromosome 4 and never observed in the control population while additional 10q alleles were detected in patients with clinical signs of the disease. As observed for 4q35 cis-duplications, the absence of uniformity within each class of genomic variants argues against the existence of founder alleles and underlines the propensity of recombination of these two subtelomeric regions. In this group, none of the patients tested is mutated for SMCHD1 or displayed a marked hypomethylation suggestive of a mutation in the DNMT3B gene. Strikingly, the vast majority of them (7 out of 11) carries a very short (1–3 units) 10q allele and a 4q chromosome with more than 20 units escaping the recent definition of FSHD2 proposed as a subclass of patients carrying between 8–20 repeated units, with cis-duplication carriers included in this subgroup based on the size of the most proximal D4Z4 array and regardless of the size of the proximal one.37 Thus, if the definition of FSHD2 as individuals carrying 8–20 repeats can be applied to many patients, recent examples including those previously described25 or those reported here (figure 1) indicate that a significant number of patients clinically diagnosed with FSHD cannot be classified in this way or might be discarded if this restrictive classification is applied. Interestingly, in our cohort of patients comprising 426 cases clinically diagnosed with FSHD, the percentage of patients carrying a mutation in SMCHD1 is close to the percentage of patients in whom we found an additional 10q allele and lower than the total percentage of patients with atypical genomic features (table 1). Furthermore, all atypical cases carry 4qAS or 4qAL alleles, with a higher proportion of 4qAS alleles, including in patients carrying very large D4Z4 arrays (B2016-2473, B2015-0851, B2013-1393, table 2) and severely affected.
Given the heterogeneity of subtelomeric regions between individuals, assembly remains incomplete, limiting comprehensive analyses. This observation is perfectly illustrated by our analyses of a large number of 4q and 10q alleles by MC revealing the large size variability among alleles, including in the proximal region corresponding to the different SSLPs and deleted in a few patients. Interestingly since differences in telomere lengths of different haplotypes have been observed,38–40 this opens a broad field for further investigations for telomere biology and diseases linked to subtelomeric imbalance. Visualisation by MC can thus be considered as a valuable tool for improving the quality of genome assembly in complex regions together with other sequencing methodologies.1 41 42 Overall, the in-depth analysis of 4qter and 10qter regions in a very large group of samples emphasises the complexity of these subtelomeric loci.
More importantly, our work also highlights the wide heterogeneity in the molecular signature of FSHD and the difficulty of interpretation of the molecular data in a significant proportion of cases, especially for genetic counselling or prenatal testing.25 26 43
Acknowledgments
The authors thank all families and patients for participating in this study.
References
Footnotes
KN and NB contributed equally.
Contributors KN designed the study, supervised, conducted, analysed the combing experiments, and conducted a survey of the data presented. NB analysed the molecular combing data. SR performed and analysed whole-exome sequencing. JDR performed and analysed the pLAM assays. CV, CC and LG conducted and analysed Southern blots and molecular combing. AM, JAU, RB, CB, AD, BE, MF, VM, SS, VT, FZ, JMC, ESC and SA provided and clinically evaluated patients. RB analysed the MC data, conducted a survey and edited the manuscript. NL designed the study and edited the manuscript. FM analysed the data, wrote, edited and submitted the manuscript. KN and FM are responsible for the overall content as guarantor of the data presented.
Funding This study was funded by Association Française contre les Myopathies (AFM grantsNMDecrypt and TRIM-RD program) and Agence Nationale de la Recherche (ANR,FSHDecipher, ANR-13-BSV1-0001).
Competing interests A patent application (No. EP08165310.7) on molecular combing for the diagnosis of FSHD1 and exploration of D4Z4 has been registered by Genomic Vision, University of the Mediterranean, and Public Assistance of the Hospitals of Marseille. NL is a co-inventor of the patent.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.