Article Text

Abstract
Background Copa syndrome is a rare autosomal dominant disorder with abnormal intracellular vesicle trafficking. The objective of this work is to expand the knowledge about this disorder by delineating phenotypic features of an unreported COPA family.
Methods and results A heterozygous missense variant (c.698 G>A, p.Arg233His) in COPA was identified in four members of a three-generation kindred with lung, autoimmune and malignant disease of unknown aetiology. Ages of onset were 56, 26, 16 and 1 year, with earlier age of onset in successive generations. Presenting symptoms were cough and dyspnoea. Findings included small lung cysts, follicular bronchiolitis, interstitial lung disease, neuroendocrine cell hyperplasia, rheumatoid arthritis, avascular necrosis and select abnormal autoimmune serologies. Neither alveolar haemorrhage nor glomerular disease were present. Features not previously associated with Copa syndrome included neuromyelitis optica, pulmonary carcinoid tumour, clear cell renal carcinoma, renal cysts, hepatic cysts, nephrolithiasis, pyelonephritis and meningitis. Longitudinal evaluations demonstrated slow progression of lung disease and extrapulmonary cysts.
Conclusions Worsening severity with successive generations may be observed in Copa syndrome. Extrapulmonary cysts, malignancies, autoimmune neurological disorders and infections are clinical features that may be associated with Copa syndrome. Further studies are indicated to fully define the phenotypic spectrum of this disorder.
- arthritis
- follicular bronchiolitis
- lung cysts
- neuromyelitis optica
- renal cell carcinoma
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Copa syndrome (MIM: 616414) is a multisystemic autosomal dominant disorder with incomplete penetrance characterised by dysfunctional cellular trafficking; it primarily involves the lungs, kidneys and joints.1 Alveolar haemorrhage, a major pulmonary feature of Copa syndrome, is associated with interstitial lung disease, follicular bronchiolitis and lung cysts.2 Renal involvement includes glomerular disease with or without immune complex deposition. Proteinuria and reduced renal function have been observed.3 Humoral autoimmunity and inflammatory polyarticular arthritis involving the knees and small joint of the hands are other manifestations.1 3 Most previously described patients presented before the sixth year of life. Patients are generally treated with immunosuppressants; some with severe pulmonary disease underwent lung transplantation.2–4
COPA encodes the alpha subunit of coatomer complex-I (COPI), which functions in the retrograde trafficking of proteins from the Golgi to the endoplasmic reticulum (ER).5 6 Nine previous kindreds have been reported with missense variants mapping to the WD40 domain of the COPA protein, along with two other reports.1 7 8 Four COPA variants in this highly conserved region are predicted to be deleterious.1 Investigations into pathogenesis of disease showed cellular dysfunction with normal levels of COPA transcript and COPA protein in cells from patients with Copa syndrome.1
We report two male and two female patients in a new kindred with Copa syndrome and show earlier age of onset of symptomatic lung disease in three successive generations. Exome analysis identified a missense variant in the COPA WD40 domain. In addition, there are atypical manifestations in these four affected patients. The clinical presentation and natural history for this kindred expand the phenotype of Copa syndrome.
Materials and methods
Patient consent and ethics approval
Written informed consent was obtained. Patients were enrolled in clinical protocols 95 H-0186 (Clinical Trials NCT00001465), 96 H-0100 (Clinical Trials NCT00001532) and/or 76-HG-0238 (Clinical Trials NCT00369421). The patients were enrolled in the National Institute of Health (NIH) Undiagnosed Diseases Programme.9–11 Clinical evaluations were performed at the NIH Clinical Centre in Bethesda, Maryland, USA.
Pulmonary function testing
Forced vital capacity, forced expiratory volume in 1 s, total lung capacity and diffusion capacity were measured (Vmax Encore, Vyaire Medical, Yorba Linda, California, USA).12
Clinical imaging
Conventional and high-resolution chest CT scans, abdominal CT scans and abdominal and brain MRI were performed.13–15
Histopathology
Tissue specimens from the proband and her father were obtained by clinically indicated open lung biopsies. Kidney tissue from the proband’s father was procured from his clinically indicated renal mass resection. Sections were stained with H&E; immunohistochemistry for synaptophysin was performed on lung tissue from the proband’s father.
Genetic analysis
Genomic DNA was isolated from peripheral blood of family members evaluated at the initial NIH admission. Three affected adult patients and the unaffected proband’s mother were analysed by whole exome sequencing.16 A heterozygous variant (c.698 G>A, p.Arg233His) in COPA segregated with affected status. Sanger sequencing confirmed this variant in these patients and the proband’s son (GeneDx, Gaithersburg, Maryland, USA).
Results
Clinical manifestations of disease
Four affected patients in three successive generations were studied (figure 1A). The proband presented at 16 years of age with cough, wheezing and chest pain. She was diagnosed with hypersensitivity pneumonitis, unresponsive to corticosteroid therapy. Dyspnoea on exertion developed at 18 years of age. At 20 years of age, nephrolithiasis was detected, and a CT scan revealed cystic lung lesions. She was referred to the NIH Clinical Center with a presumptive diagnosis of lymphangioleiomyomatosis (LAM) at 21 years of age; lung function testing revealed restriction and impaired diffusion capacity. Rheumatoid factor, antimyeloperoxidase antibody, antinuclear antigen, sedimentation rate and C-reactive protein were normal. Serum cystatin-C and beta-2 microglobulin were mildly elevated. Blood urea nitrogen, serum creatinine, urinalysis and 24 hours creatinine clearance were normal.

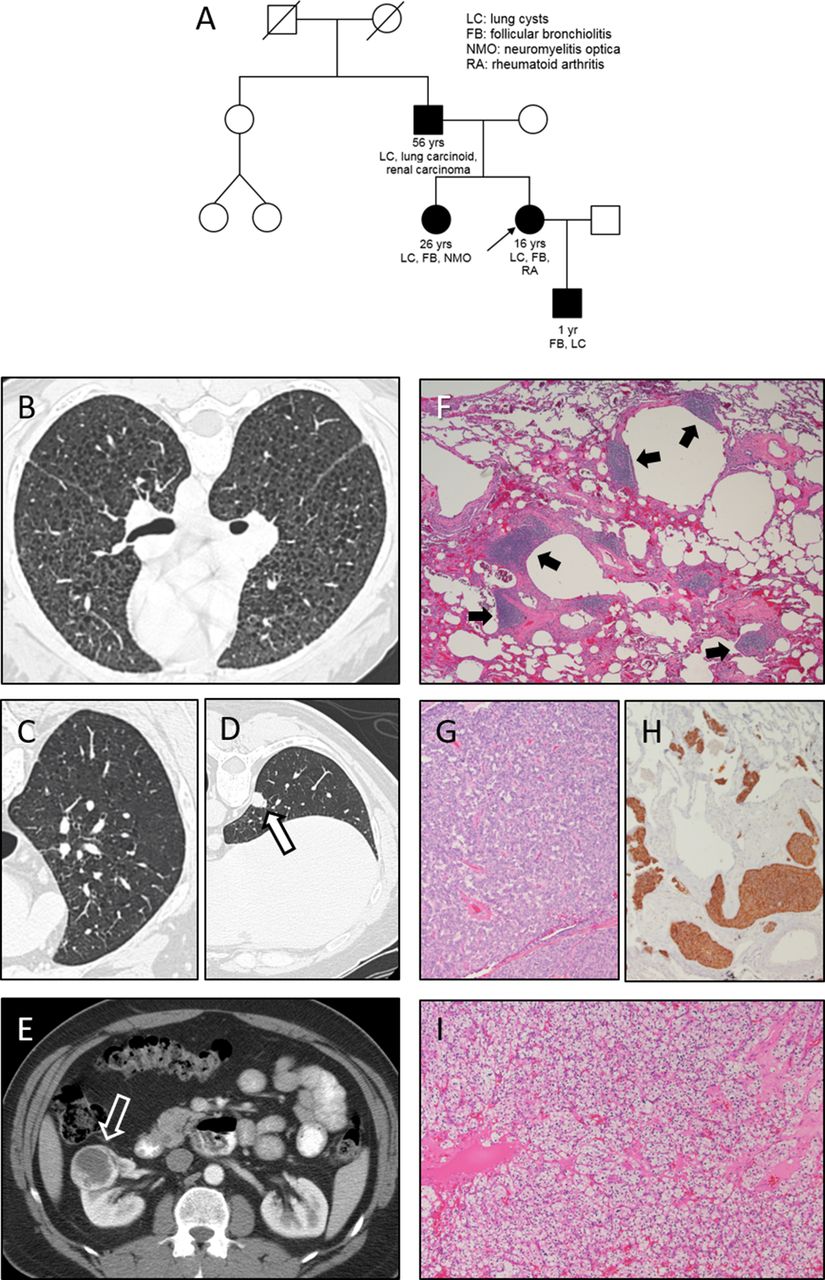{kind=link}
Pedigree indicates ages of onset of disease and the relationship between the proband (small black arrow), three affected relatives and four unaffected relatives (mother, aunt and two cousins) (A). Initial high-resolution chest CT scan of the proband and (B) shows multiple bilateral cystic lung lesions. Baseline high-resolution CT scan images demonstrate several small cystic pulmonary lesions (C) and a subpleural lung nodule (open black arrow) (D) in the proband’s asymptomatic father. A necrotic right kidney mass (open white arrow) is found by abdominal CT scan in the proband’s father (E). Lung biopsy of the proband shows reactive lymphoid follicles (solid black arrows) in a predominantly peribronchovascular distribution (F) (H&E, 4× magnification). Carcinoid tumour (G) in a resected right lower lobe nodule and neuroendocrine hyperplasia with positive immunoreactivity for synaptophysin (brown) in lung tissue (H) (10× magnification) are present in the proband’s father, who was also diagnosed with clear cell renal carcinoma (I) (H&E, 10× magnification).
The sister had hearing loss due to bacterial meningitis at 2 years of age and bilateral recurrent neuromyelitis optica starting at age 6. She was evaluated at the NIH Clinical Center at 26 years of age with dyspnoea on exertion and a presumptive diagnosis of LAM. Chest CT scan showed lung cysts. Her baseline evaluation showed restriction and severely reduced diffusion capacity. Elevated serum antimyeloperoxidase antibody, cystatin-C and beta-2 microglobulin levels were measured. Urine sediment showed 26 red blood cells/μL (normal <15) without casts. Rheumatoid factor, antinuclear antigen, sedimentation rate, C-reactive protein, blood urea nitrogen, serum creatinine, urine protein and 24 hours creatinine clearance were normal.
The proband’s 56-year-old father was asymptomatic. Given his daughters’ lung disease, an inherited disorder was considered, and he was carefully phenotyped to assign affected/unaffected status. His evaluation revealed normal lung volumes, but mildly reduced diffusion capacity. Antimyeloperoxidase antibody, antinuclear antigen, sedimentation rate, C-reactive protein, urinalysis and creatinine clearance were within normal limits. Rheumatoid factor, serum cystatin C and serum beta-2 microglobulin were mildly increased.
The proband’s son was not part of the original NIH evaluation. He presented with intermittent severe coughing paroxysms throughout childhood following respiratory syncytial virus infection at 1 year of age. At 7 years of age, chest CT revealed diffuse tiny lung nodules with a ‘tree-in-bud’ appearance and a small lung cyst. He was confirmed to have the familial COPA gene variant and was subsequently evaluated at the NIH Clinical Center. Lung function testing revealed mildly reduced diffusion capacity. Serologies were normal. Serum cystatin C and beta-2 microglobulin were slightly elevated. Urine was negative for protein, red blood cells and casts.
Imaging findings
Chest CT scans were performed during the NIH Clinical Center evaluations. The proband and sister showed a diffuse interstitial infiltrative process and scattered cystic lesions not characteristic of LAM (figure 1B). Other imaging studies failed to demonstrate any of the extrapulmonary manifestations of LAM. The proband’s renal ultrasound revealed a right renal cyst. Chest CT scans of the asymptomatic father demonstrated discrete cystic lung lesions and several lung nodules suspicious for malignancy (figure 1C and D). The proband’s son’s CT scan at 7 years of age showed a diffuse interstitial pattern and a lung cyst. A necrotic right kidney lesion was interpreted as an incidental finding in the proband’s father (figure 1E).
Histopathology of lung and kidney tissue
The proband’s lung biopsy showed findings consistent with follicular bronchiolitis. Numerous reactive lymphoid follicles were localised within peribronchovascular and subpleural regions (figure 1F). Emphysema and collections of foamy histiocytes were present. Alveolar haemorrhage was not found in the histological sections.
The father underwent lung biopsy and lung nodule resection. Pathology showed respiratory bronchiolitis, diffuse lymphoid aggregates, emphysema, neuroendocrine hyperplasia and a carcinoid tumour (figure 1G and H). He also underwent a partial nephrectomy. Histopathology revealed clear cell renal carcinoma (figure 1I). Examination of surrounding non-malignant renal tissue did not demonstrate glomerular disease.
Genetic testing
Agnostic exome sequencing of the proband, sister and both parents identified a heterozygous variant (c.698 G>A, p.Arg233His) in COPA in the father and two offspring; this was Clinical Laboratory Improvement Amendments confirmed by Sanger sequencing. Targeted analysis of DNA from the son revealed the same heterozygous variant in COPA. Sanger sequencing for this variant was negative in three unaffected relatives (an aunt and two first cousins).
Longitudinal clinical evaluations
The adult patients underwent serial evaluations over 11 years at the NIH Clinical Center. The proband’s dyspnoea slowly progressed and hypoxaemia developed at 23 years of age. Longitudinal data showed gradual decline of lung function. Chest CT scans showed progression in the appearance of innumerable small lung cysts, onset of fibrosis, lung nodule development and progressive multiple mediastinal lymph node enlargement. Abdominal imaging revealed increased size and number of renal cysts. Avascular necrosis of her femur and tibia were diagnosed at 28 years of age. Rheumatoid arthritis manifested at 29 years of age. C-reactive protein and rheumatoid factor increased to 5 mg/mL (normal 0–4.99 mg/L) and 98 IU/mL (normal <20 IU/mL), respectively; antinuclear antibody remained negative. Acute pyelonephritis occurred at 32 years of age.
The older sister has remained clinically stable, but lung function tests have demonstrated onset of airflow obstruction, and chest imaging revealed diffuse bronchial wall thickening and a new small lung nodule. Neuromyelitis optica with severe visual deficit was unchanged in the sister.
The father, who was initially asymptomatic, developed intermittent cough. Serial pulmonary function testing demonstrated gradual development of lung restriction with stable diffusion capacity. Chest CT scans showed innumerable punctate lung cysts, multiple pulmonary nodules and diffuse bronchial wall thickening. Abdominal MRI detected hepatic cysts.
Discussion
Previous cases affected by Copa syndrome provided the initial insights into the phenotypic pleiotropy of this rare autosomal dominant disorder (table 1).1 4 7 8 17 The current kindred contains individuals with some typical features of the disease, including lung cysts, follicular bronchiolitis, interstitial lung disease, neuroendocrine cell hyperplasia, positive autoimmune serologies and arthritis (table 1). Avascular necrosis, which was reported in another patient with Copa syndrome, also developed in the proband.1 However, our cohort did not have alveolar haemorrhage or nephritis, which were major elements of previously reported Copa syndrome cases.1 2
Clinical manifestations in patients with COPA variants
The presented kindred exhibited some new/atypical manifestations (table 1). One affected patient had severe vision loss due to neuromyelitis optica, an autoimmune disorder associated with autoantibodies against aquaporin-4.18 Extrapulmonary cysts in the liver and kidney, nephrolithiasis, infection (ie, acute pyelonephritis, meningitis, respiratory syncytial virus) and malignancies (ie, carcinoid tumour, renal cell carcinoma) were diagnosed in at least one affected patient; these have not been reported in patients with Copa syndrome. Cancers are known to occur in patients with various autoimmune disorders.19 20 It is unclear whether these atypical manifestations in this family are caused by or merely associated with the COPA variant.
An interesting feature of this kindred is the progressively earlier age of onset of lung disease in three consecutive generations. This phenomenon has not been reported in Copa syndrome. Symptoms first manifested in the proband at 16 years of age. Her father was asymptomatic at 56 years of age, and her son presented with symptoms at 1 year of age. A kindred with a different COPA variant includes a 32-year-old female proband and her two affected children.4 Notably, these three patients with Copa syndrome were diagnosed with follicular bronchiolitis, and their two-generation pedigree also displayed progressively more severe disease with consecutive generations. The biological basis of this phenomenon in an autosomal dominant disorder with a missense variant is unclear. It may be attributed, at least in part, to a high index of suspicion of disease and evaluation of less severely affected relatives. Technological advances with each generation could also facilitate diagnostic capabilities.
Six missense variants mapping to amino acid residues 230–243 in the WD40 region in COPA have been reported in patients of various ethnicities (table 1). Pathogenic COPA variants are associated with a defect in protein binding and retrograde protein trafficking from the Golgi to the ER.1 Analysis of tissue and cells from patients with Copa syndrome demonstrate increased ER stress, impaired autophagy, cytokine upregulation and type I interferon pathway activation.1 17 It has not been determined how cellular dysfunction in Copa syndrome contributes to disease pathogenesis.
Thirty-two individuals with Copa syndrome, including four patients in this family, are reported, and it is unclear whether there is a genotype–phenotype association.1 4 7 8 17 An Arg233His variant was found in four kindreds with 13 affected patients. Longitudinal analysis for our patients demonstrated slowly progressive cystic lung disease and follicular bronchiolitis. In contrast, follicular bronchiolitis in a 32-year-old woman and her 11-year-old son with a Glu241Lys variant progressed, and they received lung transplants.4 Although the Glu241Lys variant is associated with severe lung disease, it has not been reported in patients with Copa syndrome and kidney disease.
In conclusion, this family with Copa syndrome includes four affected individuals who are heterozygous for a missense COPA variant. This family has manifestations not reported in other patients with Copa syndrome, including neuromyelitis optica, extrapulmonary cysts and renal and neuroendocrine malignancies. Furthermore, the slow progression of follicular bronchitis differs from the more rapidly progressive disease observed in another kindred with a different COPA variant, which suggests that there may be variable expression associated with Copa syndrome. Overall, the identification of unreported clinical features in this family with Copa syndrome provides new insights into pathogenic mechanisms and hints that the phenotypic spectrum of this disorder is not yet fully defined.
Supplemental material
Acknowledgments
The authors thank our patients for their participation in our studies.
References
Footnotes
BRG and JM contributed equally.
AMT-DS and TCM contributed equally.
Contributors All authors contributed to the acquisition, analysis or interpretation of data, revised the manuscript critically for important intellectual content and approved the version to be published. AMT-DS, TCM, WAG, BRG and JM contributed to the conception of this work. AMT-DS, TCM, BRG and JM drafted the manuscript and are responsible for the overall content as guarantor(s).
Funding This research was supported by the Intramural Research Programs of the National Heart, Lung and Blood Institute, the Office of the Director, the National Cancer Institute and the National Human Genome Research Institute, National Institutes of Health.
Competing interests None declared.
Patient consent Obtained.
Ethics approval National Heart, Lung, and Blood Institute or National Human Genome Research Institute.
Provenance and peer review Not commissioned; externally peer reviewed.