Article Text

Abstract
Background Early detection of lung cancer to allow curative treatment remains challenging. Cell-free circulating tumour (ct) DNA (ctDNA) analysis may aid in malignancy assessment and early cancer diagnosis of lung nodules found in screening imagery.
Methods The multicentre clinical study enrolled 192 patients with operable occupying lung diseases. Plasma ctDNA, white cell count genomic DNA (gDNA) and tumour tissue gDNA of each patient were analysed by ultra-deep sequencing to an average of 35 000× of the coding regions of 65 lung cancer-related genes.
Results The cohort consists of a quarter of benign lung diseases and three quarters of cancer patients with all histopathology subtypes. 64% of the cancer patients are at stage I. Gene mutations detection in tissue gDNA and plasma ctDNA results in a sensitivity of 91% and specificity of 88%. When ctDNA assay was used as the test, the sensitivity was 69% and specificity 96%. As for the lung cancer patients, the assay detected 63%, 83%, 94% and 100%, for stages I, II, III and IV, respectively. In a linear discriminant analysis, combination of ctDNA, patient age and a panel of serum biomarkers boosted the overall sensitivity to 80% at a specificity of 99%. 29 out of the 65 genes harboured mutations in the patients with lung cancer with the largest number found in TP53 (30% plasma and 62% tumour tissue samples) and EGFR (20% and 40%, respectively).
Conclusion Plasma ctDNA was analysed in lung nodule assessment and early cancer detection, while an algorithm combining clinical information enhanced the test performance.
Trial registration number NCT03081741.
- circulating tumor DNA
- targeted gene NGS sequencing
- liquid biopsy
- lung nodule malignancy
- lung cancer detection
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- circulating tumor DNA
- targeted gene NGS sequencing
- liquid biopsy
- lung nodule malignancy
- lung cancer detection
Introduction
Lung cancer is the leading cause of cancer-related deaths, accounting for an estimated 1.6 million deaths each year globally.1 The prognosis of lung cancer is dependent on the stage of diagnosis, with 5-year overall survival rate decreasing dramatically from stage IA (85%) to stage IV disease (6%).2 3 It is clear that to screen and diagnose lung cancer earlier will save lives.3
Current method for lung cancer screening is low-dose computed tomography (LDCT).4 The National Lung Screening Trial demonstrated a 20% reduction in lung cancer mortality for LDCT compared with X-ray and a 6.7% all-cause mortality reduction.5 However, the imagery technique often results in indeterminate nodules6 and the false positive results lead to unnecessary invasive diagnostic procedures and increased deaths from avoidable surgeries.7
Traditionally, biopsy is used to determine malignancy of lung nodules. This approach has significant limitations as being difficult or even impossible. Molecular tests using pervasive biofluid samples, so-called liquid biopsy, are promising and urgently needed in the thoracic clinic, as demonstrated in thyroid nodules assessment where a number of molecular tests are available.8 In pulmonary nodule malignancy assessment, there is also the report of blood proteomics biomarkers by the PANOPTIC team.9
In theory, circulating tumour (ct) DNA (ctDNA) is exquisitely specific for an individual’s tumour as by definition somatic mutations are identified by their presence in tumour DNA and absence in matched normal DNA. This bypasses the issues related to the false-positivity encountered with other biomarkers, such as protein biomarkers. This specific and promising early detection method has garnered tremendous attention for cancer in general and lung in particular.10 11 The growing interest has drawn attention from international societies such as the International Association of the Study of Lung Cancer (IASLC) that has issued statement regarding liquid biopsy in the management of non-small cell lung cancer (NSCLC).12
Early detection of cancer by blood test was shown possible even with microscopic tumour proceeding radiography.13 Most recently, a preliminary analysis of early to mid-stage (stages I–III) patients with lung cancer as part of the Circulating Cancer Genome Atlas (CCGA) pan-cancer study was reported.14
In addition to mutation analysis, ctDNA epigenetics has also been evaluated and researched for early cancer detection. The studies examined DNA methylation regulation15 or the hypermethylation of the promoter regions of genes16 for the potential biomarkers of lung cancer detection.
In this study, we report a multicentre clinical trial result on genetic alterations in patients that undergo surgical resection with either benign nodules or early to midstages lung cancer. By using ultra-deep next generation sequencing (NGS) sequencing to detect very low-frequency ctDNA in a background of mostly non-tumour-derived cfDNA and comparing with the tumour tissue, we develop an assay to distinguish benign versus malignant lung lesions and detect the early cancer pathogenesis.
Methods
Patients and clinical data
The study design of the clinical trial (ClinicalTrial.gov) is prospective, cross-sectional, longitudinal and observational. It is conducted at four tier A hospitals in China to recruit patients with lung occupying diseases identified by imaging evaluation to be treated by surgery. Briefly, consecutive patients with benign lung nodules or cancers of stages I–III planned for surgical resection were assessed for eligibility, and those met the criteria (online supplementary table S1) were approached by the study team for consenting. The ones that signed the informed consent form were enrolled, and biological samples (two tubes of 10 mL peripheral blood prior to surgery collected in a cell-free DNA [cfDNA] BCT blood collection tube [Streck, Omaha, Nebraska, USA] and 10 slides of formalin-fixed paraffin-embedded (FFPE) tissues from surgery) and related clinical data including serum biomarkers were collected.
Supplementary file 1
Basic demographic and clinical data were collected using the IRB-approved clinical protocol and case report form. The initial discovery of the space occupying lesions was done through thoracic CT scan, and the read out was provided by the radiologists at the clinical site of the hospitals. The final diagnosis of the malignancy status of the nodules and cancer TNM staging was established by the pathologists of the hospital using the resected tissues.
Sequencing analysis
The above-collected DNA samples were analysed by our proprietary Sec-Seq technique as described previously.17 Briefly, blood sample were processed to separate the plasma from blood cells by centrifugation. cfDNA, gDNA of white cell count (WCC) and FFPE were extracted using QIAamp Circulating Nucleic Aid Kit, DNA mini kit, DNA FFPE Tissue kit, respectively (Qiagen, Hilden, Germany). The concentration of extracted DNA was measured using Qubit 3.0 dsDNA high-sensitivity assay (Life Technologies, Carlsbad, California, USA).
Capture probes were designed for 65 cancer-associated genes covering 241 kb genomic regions (online supplementary table S2) and synthesised by IDT (Michigan, USA). Indexed libraries were constructed using KAPA HyperPlus Kit (KK8514). Barcoding was employed to reduce noise. Postcapture multiplexed libraries were amplified with Illumina backbone primers for 16 cycles of PCR using 1× KAPA HiFi Hot Start Ready Mix and sequenced on Illumina NovaSeq platform (Illumina, California, USA) at Novogen (Nanjing, China).
Supplementary file 2
Horizon’s Partners Spike-in control (Horizon, Cambridge, UK) was used in a serial dilution (from 0.0005 to 1) using the wild-type reference genome and the provided reference standard. Reference variants include EGFR (L858R and T790M), KRAS (G12D), NRAS (Q61K and A59T) and PIK3CA (E545K) or NA12878 (12 mutations) and NA24385 (29 mutations).
Bioinformatics data analysis
Reads with quality score <30 or having >5% of positions differ from the rest of the reads targeting the same region were removed. The results were then mapped to the human reference genome (hg19) using BWA (V.0.7.15-r1140). We used the start mapping positions, the length and the dual barcode on both side of merged paired-end fragments to form reads groups amplified from every primary cfDNA molecules and to identify incorrect base produced due to PCR errors.
Variant calling for single nucleotide variation or insertion/deletion was performed using samtools mpileup tool (V.1.3.1). For ctDNA samples, a variant was selected as a candidate somatic mutation when: (1) two distinct paired reads (each redundantly sequenced at least three times) contained the mutation, (2) effective reads depth >500 (captured primary cfDNA molecules >500) and (3) the corresponding allele frequency in WCC is less than 1%.
Mutation annotation and classification
The variants were called by SnpEff (V.4.3o) and annotated by COSMIC (V.85), ExAc, ClinVar and 1000 Genome. The following variants were eliminated: (1) intergenomic or intronic (except for splicing junction); (2) synonymous; and (3) variant allele frequency (VAF) <0.2% in ctDNA or <1% in FFPE samples. Previously reported and confirmed pathogenic mutations in the clinical samples of lung cancer of all human races and ethnicities will be considered as lung cancer related.
Statistical methods
Data were summarised using descriptive statistics. Fisher’s exact test was used to compare any two subgroups. Wilcoxon rank-sum test was used to compare median age between any two subgroups (stages I, II and III) or mutant groups (mutant vs wild type).
Linear discriminant analysis (LDA) was performed on improving mutation analysis. The model considers the age of the patients, ctDNA mutations and the serum biomarkers. It was developed using the 10-fold cross-validation by dividing the samples into training and validation subsets. The test sensitivity and specificity was calculated, and the area under the receiver operator curve (AUROC) was plotted.
Results
Patient demographic and clinical analysis
In total, 192 patients with pulmonary space occupying lesions (136 malignant and 56 benign) pathologically diagnosed and surgically treated were included in this analysis. These patients were recruited from four clinical sites: Xiangya No. 2 Hospital in Changsha, Hunan Province, Beijing University Shenzhen Hospital, Shenzhen, Huizhou People’s Hospital, Huizhou, and No.2 People’s Hospital in Shenzhen, Guangdong Province.
The average age is 56.5 (range 26–79) years, and male proportion is 59%. These numbers are 50.1 (26–73) years and 55% for the benign group, 59.1 (27–79) years and 60% for lung cancer group with a statistically significant difference. No statistically significant difference in terms of smoke status (32% vs 35%) and family history (7% vs 8%) was found between the two groups (table 1).
Patient demographics
For the benign lesions, the most common diseases diagnosed are pneumonia (n=14, 25%), tuberculosis (n=12, 21%), pulmonary fibrosis (n=4, 7%) and necrosis granuloma (n=3, 5%).
Lung cancer distribution was 87, 29 and 17 in stages I, II and III, respectively. The average size of the nodules for lung cancer patients is 2.9 (range 0.5–9.0) cm, and for each subgroup: (1) stage I: 2.2 (0.5–4.0) cm; (2) stage II: 3.8 (1.0–7.0) cm; and (3) stage III: 5.0 (1.3–9.0) cm (table 2). The average size of the nodules in the benign group is 2.3 (0.3–6.0) cm, which is statistically smaller than that of the malignant group.
Lung cancer histological diagnosis and stages
Stage IV lung cancer was to be excluded according to the trial design, but due to the fact of late coming of pathological result, we have samples from three patients of this stage collected and analysed. The data for these samples are not statistically meaningful and therefore are included for completeness in the online supplementary tables but not taking into account in the result tables and figures in the main text.
All of the patients are with solitary pulmonary nodules, except for two patients who had two nodules each that are malignant.
Genetic profiling and mutation burden
For each patient, three biospecimen samples including plasma ctDNA, WCC and FFPE tumour tissue were sequenced. For ctDNA, the average sequencing depth is 35 000 with 1350 unique reads after deduplication.
In total, 312 occurrences and 274 unique somatic mutations were found in 29 genes from either plasma ctDNA or tissue DNA in 120 cancer and 5 benign cases (online supplementary table S3).
In the benign lesions, 2 out of 56 patients (3.6%) had four non-driver gene mutations in ctDNA, and three patients (5.4%) had two non-driver mutations in FFPE samples.
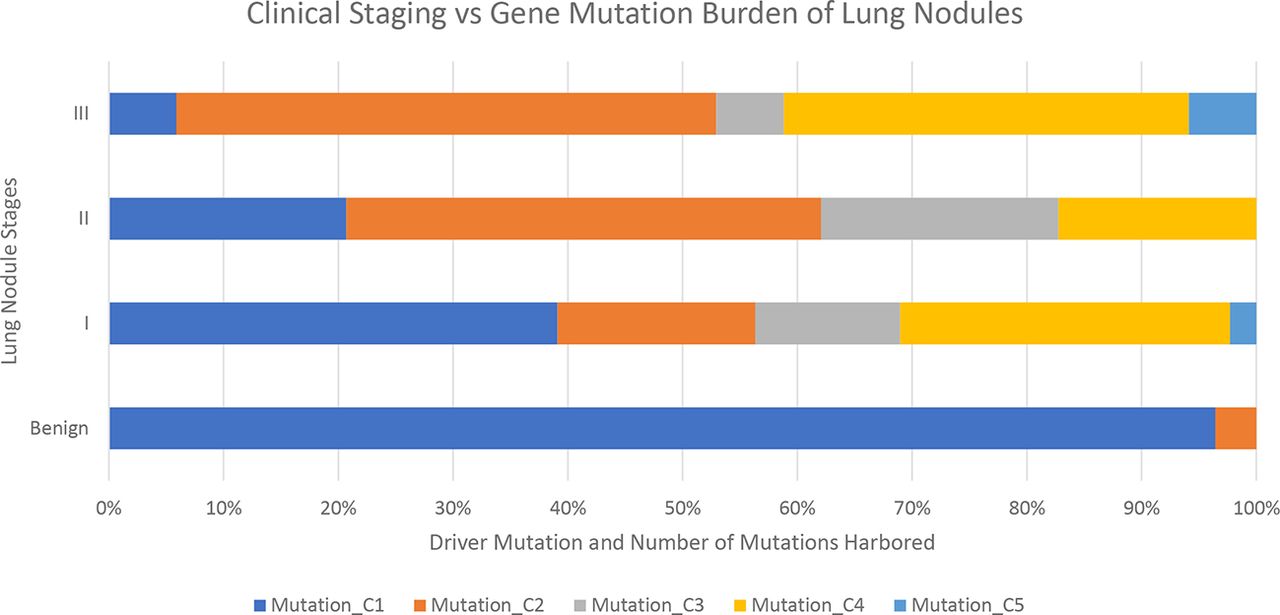
Among the lung cancer patients, 88% (120 out of 136 patients) were found to harbour at least one mutation in ctDNA or tumour tissue. When analysed by stage of cancer, the class, that is, whether driver or non-driver mutation, and number of mutations increase as the stage advances (figure 1).

Lung cancer gene mutation burden in benign and various stages of patients. Mutation types: C1: no mutation found; C2: one non-driver mutation found; C3: one driver mutation found; C4: both driver mutation and non-driver mutation found; C5: more than one non-driver mutation found.
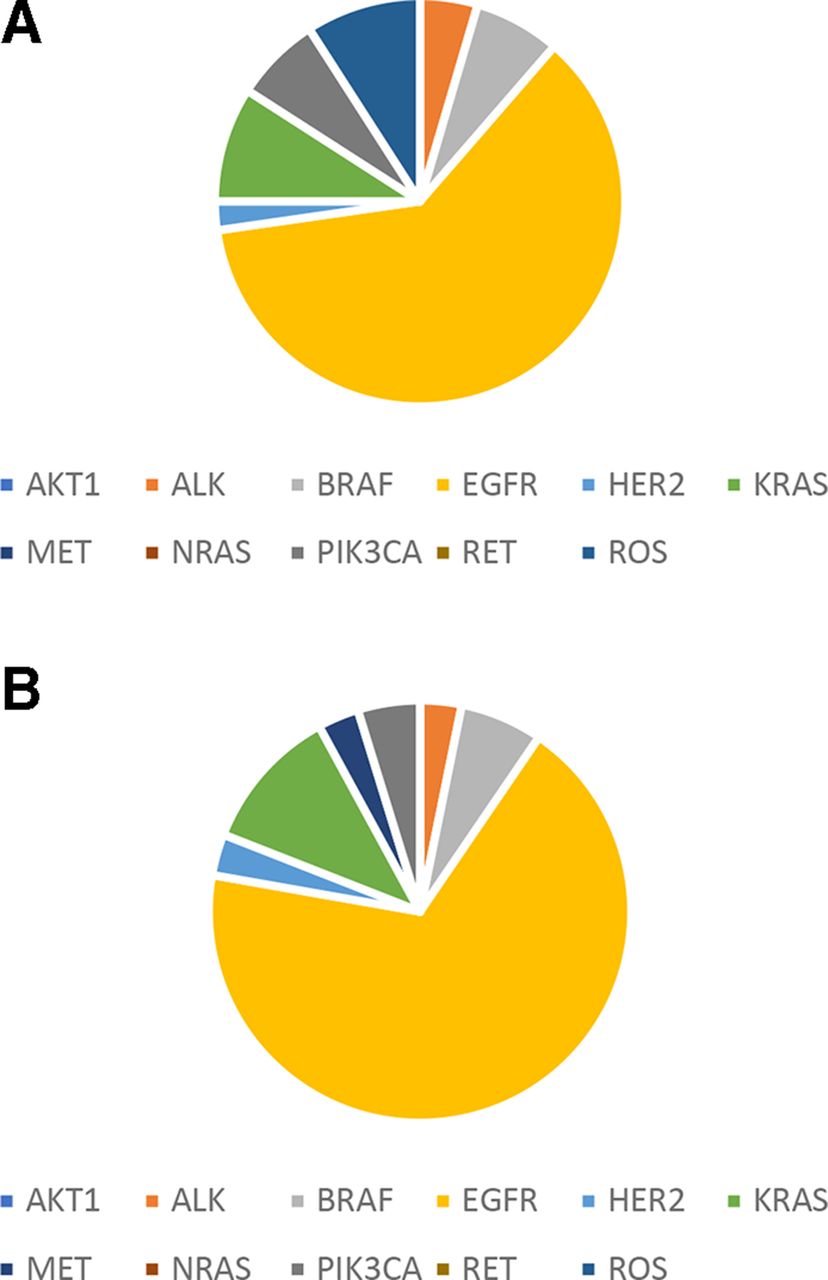
Mutations were found in 9 known lung cancer genes of ALK, BRAF, EGFR, HER2, KRAS, MET, NRAS, PIK3CA and ROS1 (figure 2) out of the 12 genes defined as drivers for lung cancer.18 The most commonly mutated genes in the patients with lung cancer are TP53 (44%) and EGFR (35%) (online supplementary figure S1).
Supplementary file 3
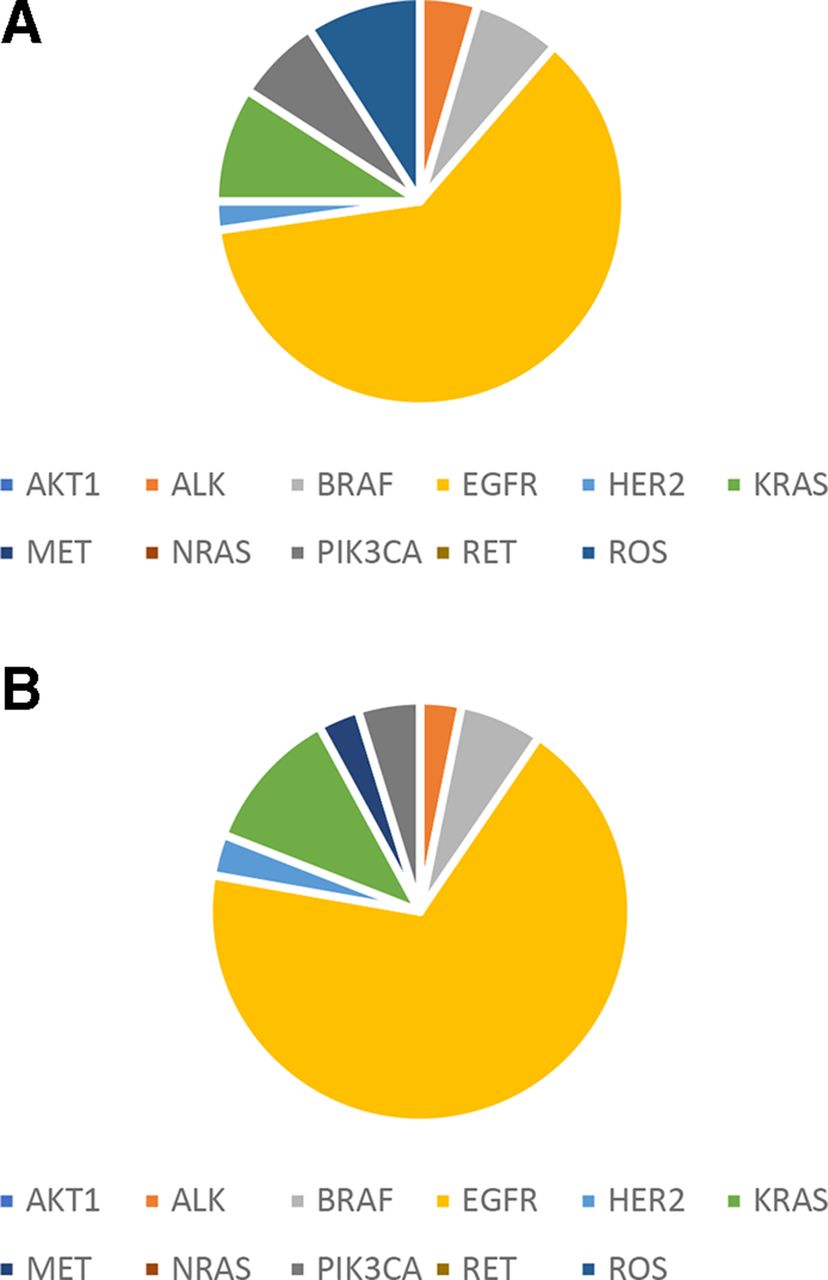
Driver mutation distribution in patients with lung cancer patients. (A) Plasma ctDNA. (B) FFPE gDNA. ctDNA, circulating tumour DNA; gDNA, genomic DNA.
The most commonly occurred mutation is EGFR L858R (found in 24 samples) followed by EGFR exon 19 deletion (in 15 samples). The largest number of mutations was found in a patient who harboured 15 mutations.
Concordance between ctDNA and tumour tissue
Concordance is defined as at least one gene mutation is the same in both the plasma ctDNA and the FFPE tumour tissue gDNA of a patient. When completely no mutations were found in both the blood ctDNA and tissue gDNA, it is also considered as concordant of the two samples.
Among the 136 malignant cases, the overall concordance rate is 27%. Concordance was higher in the driver genes, 46%. The shared mutation rate increases as the stage of the cancer advances: 14%, 48%, 41% and 67% at stage I, II, III, and IV, respectively (online supplementary figure S2).
Comparison with serum biomarkers
A panel of six serum protein tumour biomarkers was also analysed, which has a sensitivity of 51% with a specificity of 83% (table 3). These markers include NSE, CYFRA 21–1, CEA, ProGRP, CA-125 and SCC. When the most sensitive marker of CYFRA 21–1 was considered alone, the sensitivity was merely 25% at a specificity of 95% (table 3).
Lung nodule malignancy prediction diagnostic test performance
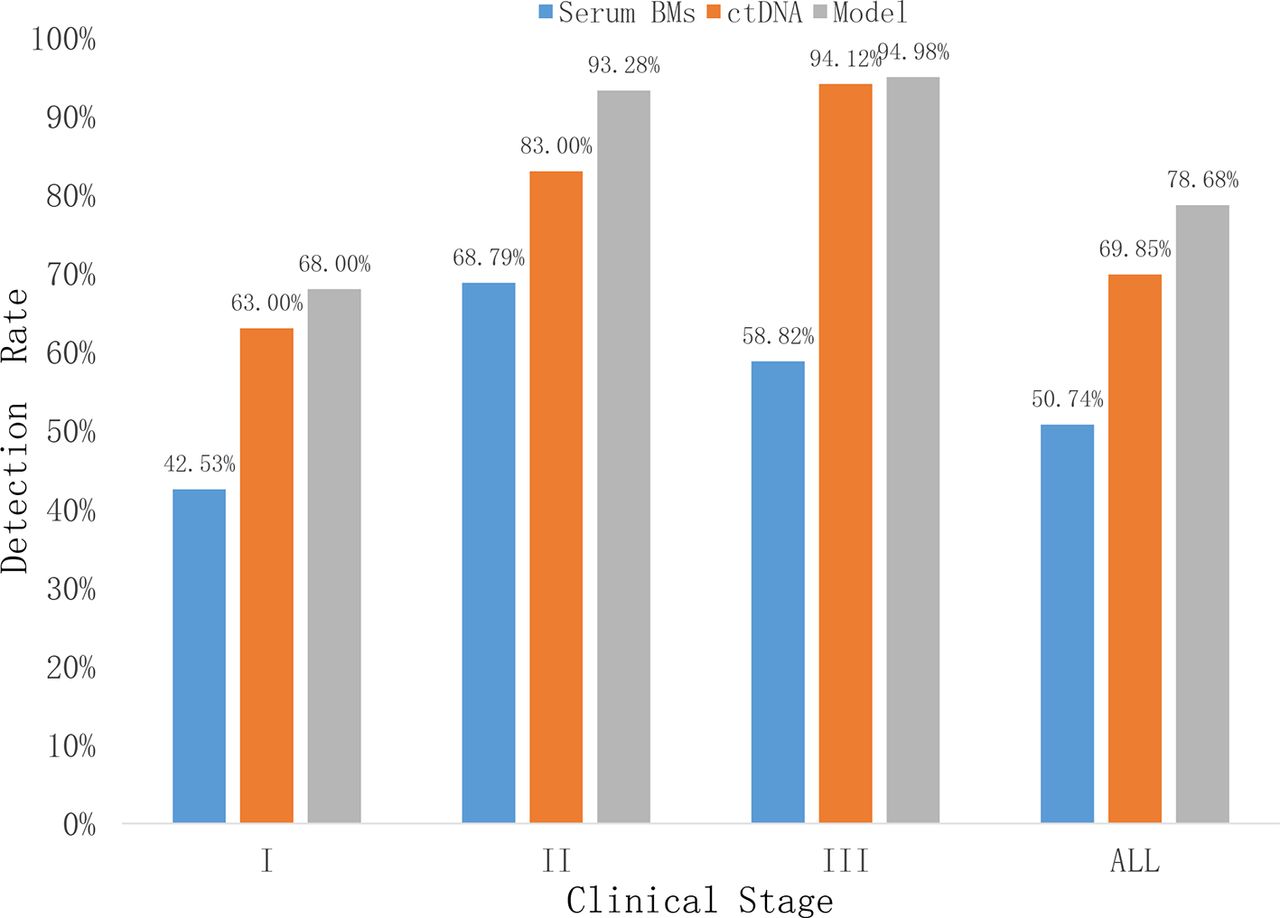
In comparison, the profiling by ctDNA showed a higher sensitivity in detecting lung cancer. The sensitivity increases as the stage advances and ctDNA outperform the serum biomarkers in all stages (figure 3).
{kind=link}
{kind=link}
{kind=link}
Lung cancer detection rate by ctDNA, serum biomarkers and LDA model per stage. Model=LDA model. ctDNA, circulating tumour DNA; LDA, linear discriminant analysis.
Linear discriminant lung cancer algorithm
The mutation profiling classifies the lung nodules from benign to malignant (figure 1). The benign samples harbour very little to no mutations. The cancer patients have increasing level of mutations both in terms of category and numbers as stage progresses. From stage I to III, there are more mutations found, and they are more often in driver genes.
For lung cancer detection, the overall sensitivity of plasma ctDNA was 69% at the specificity of 96% (table 3). According to cancer stages, the sensitivity rate is 63%, 83% and 94% for stages I, II and III, respectively (figure 3).
We further conducted an LDA where patient age, smoking status and serum protein markers were considered (online supplementary figure S2). The combined model of ctDNA mutations and serum biomarkers improved the sensitivity and specificity to 80% and 99%, respectively (table 3).
Discussion
Lung cancer screening recommended by the guidelines targets populations at high risk of developing lung cancer, such as patients aged above 55 years, heavy smokers and having chronic obstructive pulmonary disease (COPD) and with family history of lung cancer. In general, by definition of 50–500 mm3,6 screening by imagery techniques could result in about 30%–40% indeterminate nodules that need to be further evaluated. In our cohort, we have >60% nodules in this size range making it an urgent need for malignancy assessment.
Our patient population drawn from major hospitals in southern and central China has a median age of 56.5 years, just above the threshold for lung cancer screening. The clinical outcome confirmed the age of above 55 years as a risk factor for lung cancer: the median age of the patients with benign nodules (50.1 years) was about 10 years younger than those having lung cancer (median age of 59.1 years). Although not strictly a screening population, our cross-sectional cohort drawn from the consecutive patients with sign and symptom revealed pulmonary nodules of both benign or malignant nature represents a closer step towards high-risk screening.
Smoking status and family history were not significantly different between the benign and the malignant cases in our cohort. This result is somewhat uncommon given that the role of smoking in lung cancer is considered documented. However, our cohort may not be large enough to observe the effect, especially when considering our cases (early stage cancers) and controls (benign lung lesions) are not the typically studied previously.
Many potential explanations could still be explored. On one hand, our Chinese cohort has 41.1% of women, which has less of smokers. Gender distribution also corroborates: women are more in the benign (44.6%) than the cancer group (39.7%). On the other hand, the impact of smoking should be considered more broadly as an environmental factor than tobacco smoking alone. For example, the time spent on Chinese-style cooking could be a potential risk. Unfortunately, no such data were collected. As a proxy, however, the gender distribution difference could be explored since women usually spends more time in the kitchen. While we have more male patients in the cancer group (about 60%), the opposite is true for the benign group, which has less (about 55%).
On the potential interaction of smoking and genes, although there are studies about the association of smoking and tumour driving gene mutations, such as EGFR, ALK, KRAS and TP53 genes (reviewed in refs 19–22), these are usually studied in patients with late-stage cancer, while ours is heavily centred on the early stage patients (>85% for stages I and II). It is shown that histopathology, gender and ethnicity could also impact the mutation profile of smoking versus non-smoking lung cancers.19
It is reported that tumour burden of lung cancer corresponds to its size.23 Our data confirm that the size of the nodule relates to malignancy and progression. The average tumour size increases from 22 mm in stage I to 38 mm in stage II and to 50 mm in stage III (table 2). However, there is no significant difference between a benign nodule and that of the stage I cancer (tables 1 and 2).
The concordance rate between tissue and corresponding plasma ctDNA also reflects the challenge of early cancer liquid biopsy. Our study is heavy in stage I patients (64%, table 2) which has a rate of 32% and causes the overall rate of 36%. The rate increases as stage advances—up to the highest of 78% and the average of stages I–III is 53%. In CCGA, a set of 73 early to midstage (stages I–III) lung cancer samples showed a similar rate of 59% (95% CI 47% to 70%).13 In another very small study of 31 paired lung cancer tissues and plasma DNA samples with 10 000-fold ctDNA sequencing depth, the concordance of mutation between tumour tissue DNA and ctDNA was merely 3.9%.24 Ours is more like CCGA in that we both sequenced ctDNA to the depth >40 000×. Another meta-analysis has also put the pooled sensitivity in the range of 60%s.25
The gene mutations shared between the plasma ctDNA and the FFPE tumour tissue increase as the lung cancer stage advances. This is in alignment with the previous report that stage IV tumour has the highest concordance,26 as well as that as the tumour is getting larger, the amount of DNA fragments it sheds into the blood stream will increase.23 27 In regards to the cases where there is a discordance between mutations in plasma ctDNA and tissue, many factors could be contributing.
For the mutations found in tissue but not ctDNA samples, it could be due to the challenge of the weak ctDNA signal at early stage28 29 like the case of our cohort. The kinetics of ctDNA in the circulation and hence the timing of blood sampling in relation to tissue sampling may have influence that is not yet well understood. The effect of ageing induced clonal haematopoiesis or ageing clonal expansion30 could also play a role. In addition, there is report that leukocytes31harbour and release oncogenic gDNA into the blood and therefore may have impact on the detection of somatic mutations. All these issues will be further studied.
In terms of the mutations found in ctDNA but not in tumour tissue, we believe the most likely explanation is the issue of intertumour and intratumour heterogeneity.32 33 Blood sample is more homogeneous and could provide a holistic view of the genetic profile released in the plasma ctDNA by the tumour, while tissue sampling is localised to some specific clone or clones of the cancerous lesions.
Another issue to be watched for in ctDNA analysis is the noisy background of the cfDNA. The majority of variations found are from the WBC, a phenomenon called clonal hematopoiesis.34 For this reason, the ctDNA studies should include the matched WCC sequencing such as ours.
Although somatic mutations showed strong feasibility of detecting malignancy and staging the cancer by plasma ctDNA, there more factors to be considered. Therefore, combining clinical and genomic features improved the test performance as shown by our LDA modelling (table 3). Integrated classifiers have also been explored in terms of plasma protein biomarkers such as the ones tested by some panel components in our cohort.35 One study involving 60 patients with NSCLC, 40 patients with COPD and 40 healthy controls showed that combining the serum cfDNA concentrations and integrity and CEA improves sensitivity to 93.3%.36
Liquid biopsy starts moving into cancer clinics in therapy selection.37–40 A very recent cohort study of over 300 patients with advanced stage lung cancer using ctDNA and/or matched tumour tissue NGS mutation testing guidance for therapy selection showed the utility of liquid biopsy in increasing the positivity of drug selection and treatment outcome.39 Another single-centre study of 102 patients investigated the role of ctDNA in detecting driver gene or other actionable mutations for lung cancer therapy precision and resistance management including serial sampling, in the context where tissue biopsy is limited or could be unavailable.40 Another study identified 17 miRNA species in the exosomes of the blood that are differentially expressed in cancer (both NSCLC and SCLC) and controls.41
Early detection of lung cancer using blood samples is emerging, and similar level of detection performance as ours has been reported.13 14 The CCGA study reported a sensitivity of 54% and specificity of 98% for early-stage cancer detection in 127 patients with lung cancer. The potential use of ctDNA for early detection of other cancers has also been reported.42 43
There are, however, a number of limitation and challenges. First of all, the sample sizes of the early detection studies are usually small especially the number of healthy controls. Second, ctDNA amount correlates with cancer stage.44 Therefore, the consensus is that ultra-deep sequencing of 40 000× is required to detect the low-frequency mutations in the 10 mL blood. Finally, the less-than-expected driver mutation concordance between ctDNA and tumour DNA may reflect genetic heterogeneity and indicate tumour evolution45 suggesting that other types of genes and mutations should be considered as well.
Acknowledgments
The authors would like to thank the physicians and nurses in the Department of Thoracic Surgery, No. 2 Xiangya Hospital, Thoracic Department, Peking University Shenzhen Hospital, Department of Oncology, Shenzhen Second People’s Hospital, Oncology Center and Department of Thoracic Surgery, Sun Yat-sen University Cancer Center for their work in enrolling the patients, collecting biospecimen samples and related clinical data. We would like to thank the laboratory staff at Vienomics for processing the samples and conducting the sequencing procedures. Finally, but not the least, we would like to express our highest appreciation of the participation and contribution of our patients in the study.
References
Footnotes
MP, YX and XL contributed equally.
Contributors MP, YX, YQ, FCh, HY, FY and GT participated in patient recruitment; FCa, CL and CX participated in clinical trial and patient sample management; XL, XY and FX participated in genomic sequencing; XT, DK, BH and CX participated in bioinformatics data analysis; and CX, XL, FY and GT participated in manuscript writing and review.
Funding This work was supported by Innovation Fund of Shenzhen China (Grant No: CKCY2016082916544973 and Grant No: CYZZ20170406170950746); Technological Innovation Research Program of Shenzhen China (Grant No: JSGG20160428090301587 and Grant No: JSGG201704141042216477); State Key Research Program of China (Grant No: 2016YFA0501604); the Young Scientist Innovation Team Project of Hubei Colleges (Grant No: T201510); the Key Project of Health and Family Planning Commission of Hubei Province (Grant No: WJ2017Z023), Science technology and innovation committee of Shenzhen for research projects (JSGG20160428090301587) and Scientific Research Project of Shenzhen Health and Family Planning System (SZLY2017008/SZLY2018020).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Obtained.