Article Text

Abstract
Background Inherited cystic kidney disorders are a common cause of end-stage renal disease. Over 50 ciliopathy genes, which encode proteins that influence the structure and function of the primary cilia, are implicated in cystic kidney disease.
Methods To define the phenotype and genotype of cystic kidney disease in fetuses and neonates, we correlated antenatal ultrasound examination and postnatal renal ultrasound examination with targeted exon sequencing, using a renal gene panel. A cohort of 44 families in whom antenatal renal ultrasound scanning findings in affected cases included bilateral cystic kidney disease, echogenic kidneys or enlarged kidneys was investigated.
Results In this cohort, disease phenotypes were severe with 36 cases of stillbirth or perinatal death. Extra renal malformations, including encephalocele, polydactyly and heart malformations, consistent with ciliopathy phenotypes, were frequently detected. Renal gene panel testing identified causative mutations in 21 out of 34 families (62%), where patient and parental DNA was available. In the remaining 10 families, where only parental DNA was available, 7 inferred causative mutations were found. Together, mutations were found in 12 different genes with a total of 13 novel pathogenic variants, including an inferred novel variant in NEK8. Mutations in CC2D2A were the most common cause of an antenatal cystic kidney disease and a suspected ciliopathy in our cohort.
Conclusions In families with ciliopathy phenotypes, mutational analysis using a targeted renal gene panel allows a rapid molecular diagnosis and provides important information for patients, parents and their physicians.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
The formation of cysts in kidney is a disease phenotype common to many inherited human diseases.1 Kidney cysts are fluid-filled epithelial lined structures arising from dilation in any part of the nephron or collecting duct. Cystic kidney disorders are a common cause of end-stage renal disease (ESRD). It is estimated that the prevalence of cystic kidney disease is 4.81% in the Arabian Gulf countries.2
Ciliopathy syndromes are inherited syndromes that are frequently associated with cystic kidneys and to date, mutations in over 50 genes have been identified.3 These include autosomal-dominant polycystic kidney disease (ADPKD), autosomal-recessive polycystic kidney disease (ARPKD), various forms of nephronophthisis (NPHP), Joubert syndrome (JBTS), Meckel–Gruber syndrome (MKS), Bardet–Biedl syndrome (BBS) and many others.4 ADPKD is common and accounts for approximately 5–10% of the ESRD cases worldwide.5 Mutations in two genes, PKD1 (85% of patients with ADPKD) and PKD2 (15% of patients with ADPKD) underlie ADPKD.6 One to two per cent of patients with ADPKD may present as neonates with cystic kidneys.7 Biallelic mutations/variants in PKD1 and PKD2 have been described to give a severe neonatal onset of cystic kidney disease.8 ,9
ARPKD is a rarer condition affecting 1 in every 20 000 live births.10 It may be diagnosed in utero or prenatally by sonography showing bilateral large echogenic kidneys, and oligohydramnios in the most severe cases. Mutations in the polycystic kidney and hepatic disease 1 (PKHD1) gene are responsible for ARPKD, the severity of which depends on the type of mutations.11 The PKHD1 gene is located on chromosome 6p21 and encodes a fibrocystin protein that localises to the primary cilium of renal epithelial cells. There is a high risk of fetal presentation and neonatal death if the fetus carries two truncating mutations.12
Inherited ciliopathies may also cause multisystem pathology, which may be severe and result in early death for many patients. Aside from cystic kidney disease, other common clinical features of ciliopathies include hepatobiliary disease, laterality defects, polydactyly, agenesis of corpus callosum, retinal degeneration and occipital encephalocele.13 Ciliopathies with prominent renal phenotypes include NPHP, JBTS and MKS.
NPHP is an autosomal-recessive disorder responsible for 6–10% of ESRD in children.14 NPHP is characterised by cysts that are typically restricted to the corticomedullary junction region of the kidney, and the kidney size is normal or reduced.15 The disease is genetically heterogeneous. Mutations in over 20 different recessive genes (including NPHP1–NPHP19, AHI1 and XPNPEP3) have been identified in about 50% of NPHP patients.16 Infantile NPHP is a disease that progresses to ESRD usually before the age of 2 years and is characterised by cortical microcysts associated with tubulointerstitial lesions. Classically, it is linked to NPHP2/INVS gene encoding inversin, but patients carrying NPHP3 mutations may also develop the infantile phenotype frequently associated with liver involvement.17
JBTS is neurodevelopmental disorder characterised by cerebellar vermis aplasia (CVA), a significant malformation of the cerebellum that is linked to ataxia and may be seen on brain MRI as ‘molar tooth sign’.18 JBTS follows an autosomal-recessive inheritance pattern and there are currently over 26 known causative genes.19–21 JBTS may be associated with cystic renal disease in a subset of cases.
MKS is a prenatally lethal autosomal-recessive condition characterised by occipital encephalocele, bilateral renal cystic dysplasia, hepatic ductal proliferation, fibrosis, cysts and polydactyly.22 Patients with MKS invariably die from respiratory and/or renal failure. Genetic heterogeneity of MKS has been established with now 13 reported genes involved.23–25
For many of these ciliopathy syndromes, significant phenotypic variability has been observed even between members of the same family, making clinical diagnosis, prediction of clinical progression and genetic counselling a challenge.
Antenatal screening using ultrasound scanning (USS) is a means by which cystic kidney disease can be readily detected. Serial ultrasound evaluation starting from 11 weeks of gestation onwards can be used as a screening modality.26 Abnormal findings that point towards a renal ciliopathy include increased size of kidneys, a bright echotexture (hyperechogenicity) and a loss of the normal corticomedullary differentiation. Perinatal ultrasound appearance of kidneys can look similar in fetuses with ARPKD, perinatal-onset ADPKD, MKS and some forms of NPHP. In addition to renal anomalies, prenatal ultrasound can detect other features of ciliopathies such as encephalocele, polydactyly, situs inversus, agenesis of the corpus callosum, Dandy–Walker malformation, fibrosis of the liver and structural heart defects.22
In this study, we have combined antenatal ultrasound examination of the fetus and targeted molecular genetic ‘panel testing’ for inherited renal disorders to characterise a cohort of Saudi Arabian patients who presented antenatally with features of an inherited renal ciliopathy.
Materials and methods
Study cohort
The cohort consists of 44 Saudi Arabian families where there was evidence of antenatal USS anomalies of the kidney, which included cystic kidney disease, enlarged kidneys and echogenic kidneys. Additional antenatal USS findings including central nervous system (CNS) anomalies (encephalocele, CVA, ventriculomegaly), cardiac defects (congenital heart malformation, pericardial effusion) and skeletal defects (narrow thorax, polydactyly) were documented. Clinical phenotypes postnatally were also reviewed, including postnatal renal USS. For molecular genetic investigations, the cohort was divided into two groups: Group A, where DNA was available from the affected fetuses and their parents (n=34 families) and Group B, where DNA was available from both parents but not the affected child (n=10 families) (table 1). Following informed consent, DNA was extracted from available chorionic villus sampling, amniotic fluid, placental blood or peripheral blood cells using the Gentra Systems PUREGENE DNA Isolation kit (Qiagen, Valencia, California, USA). Ethical and study permissions were approved by the Research Advisory Council of King Faisal Specialist Hospital, Riyadh, Saudi Arabia (RAC#2050 045). We confirm that all the diagnostic genetic work was performed in Saudi Arabia with full ethical approval. The UK centre acted in an advisory and strategic manner to direct the study.
Clinical and molecular findings in cohort of antenatal cystic kidney disease and ciliopathy phenotypes
Antenatal USS examination
Prenatal anatomy USS examination was performed at the Obstetrics and Gynecology Department, King Faisal Specialist Hospital and Research Centre, between weeks 18 and 22 of pregnancy. For cases with a known family history of cystic kidney disease/ciliopathy serial, antenatal USS examinations started between 12 and 22 weeks. For new referrals and unknown family history, antenatal USS started at the first visit. Fetal anatomy was reported as either normal or abnormal with explanations for features that includes cranium, cerebral ventricles, posterior fossa, face, spine, chest, cardiac four-chamber view, cardiac outflow tracts, heart axis, cardiac situs, stomach, bowel, kidneys, bladder, abdominal cord insertion, number of cord vessels, upper extremities and presence of hands, and lower extremities and presence of feet. Published reference values for renal length and volume27 and for renal volumes based on three-dimensional ultrasound28 were used. Fetal death was defined as an intrauterine death greater than 10 weeks of gestation. A perinatal death is defined as a death within 7 days of birth, and an infant death is defined as a death within 1 year of birth.
Maternal cell contamination and molecular karyotyping
In all fetal DNA samples, maternal cell contamination was excluded by using the AmpFLSTR Identifiler PCR Amplification Kit as described by the manufacturer (Applied Biosystems, Life Technologies, Paisley, UK). Where available fetal DNA was used for molecular karyotyping (Affymetrix CytoScan HD Array Kit, Santa Clara, California, USA) to exclude chromosomal aneuploidy and to determine regions of homozygosity in the affected patient.
Targeted renal genes panel and next generation sequencing
A customised 90 renal genes panel that includes ciliopathy genes (including 3 polycystic kidney disease genes, 10 NPHP genes, 9 JBTS genes and 11 MKS genes) as well as and other inherited renal disorders (see online supplementary table S1) was prepared using Life Technologies proprietary AmpliSeq multiplexing assay. This panel has previously undergone validation for its analytical sensitivity and specificity using 107 renal patients and had 89% base reads on target with a read depth (base coverage) of 840 after alignment and a of 98% coverage of all genes.29 All samples were prepared within the Saudi Human Genome Project Laboratories and loaded onto a Proton I chip, and sequencing was performed on an Ion Proton system (Ion Torrent—Life Technologies) as recommended by the manufacturer and as previously described.29
NGS analysis pipeline
The analysis pipeline for processing the next generation sequencing (NGS) reads went through several steps. Reads were examined for quality and parts of reads with low-quality value were trimmed out. The reads were then aligned to the human reference genome GRCh37/Hg19 with the Torrent Mapping Alignment Program (TMAP) Aligner software. Once the reads were aligned, the variants were called using the Torrent Variant Caller (TVC) program. The TMAP and the TVC programs are distributed as part of the Torrent Suite (https://github.com/iontorrent/TS) package. The resulting variant files were stored in variant call format (VCF) files. The VCF file generated for each sample was processed through an annotation pipeline against databases such as OMIM, GenBank, dbSNP, 1000 genome project, Human Gene Mutation Database, and a local database (SGP737) of 550 patients containing Arab-specific variants. Variants with a minor allele frequency (MAF) >1% were discounted.
In addition to allele frequency, annotation provides pathogenicity scores, homozygosity/heterozygosity, read quality scores and other parameters used to identify candidate causative variants. All NGS and targeted sequencing and bioinformatics analysis were performed at the Saudi Human Genome Project Laboratories at KFSHRC and KACST.
For predicting the damaging effect of the reported mutation, four in silico prediction tools were used: PolyPhen-2 (http://genetics.bwh.harvard.edu/pph/), Provean (http://provean.jcvi.org/index.php), MutationTaster (http://www.mutationtaster.org/) and Human Splicing Finder (http://www.umd.be/HSF/#). Reported allele frequency of all putative pathogenic variants was determined using the ExAC database (http://exac.broadinstitute.org/), and evolutionary conservation was determined from sequence alignments using MutationTaster and UCSC (https://genome.ucsc.edu).
Sanger sequencing
Direct sequencing of PCR amplicons was carried out to confirm positive gene panel results. PCR was performed using Qiagen (Manchester, UK) master mix kit. Oligonucleotide primers for PCR amplification of targeted genomic DNA were designed using Primer3 software (http://frodo.wi.mit.edu/) and synthesised by Metabion International AG (Munich, Germany). Primer sequences are available on request. Following treatment with the Agencourt AMPure PCR purification system (Agencourt Bioscience, Beverly, Massachusetts, USA), products were sequenced using BigDye Terminator Cycle Sequencing kit (PE Applied Biosystems, Massachusetts, USA) and run on an ABI 3730xl capillary sequencer. Sequences were analysed using Mutation Surveyor software V.3.24 (SoftGenetics LLC, State College, Pennsylvania, USA).
Results and discussion
A cohort of 44 families were analysed, where 38 (86%) were known to be consanguineous and 26 (59%) had more than one affected fetus. The antenatal renal USS findings included either bilateral cystic kidney disease, echogenic kidneys or enlarged kidneys in all cases (table 1). Antenatal USS also detected extrarenal malformations at a high rate (figure 1): 25 (57%) had oligohydramnios or anhydramnios, 14 (32%) had encephalocele and 9 (20%) had CVA. Other anomalies included limb defects including polydactyly and structural cardiac defects. The phenotype of this cohort was extremely severe with 38 (86%) cases dying as stillborn infants or perinatally. Only six cases survived the perinatal period (table 1).
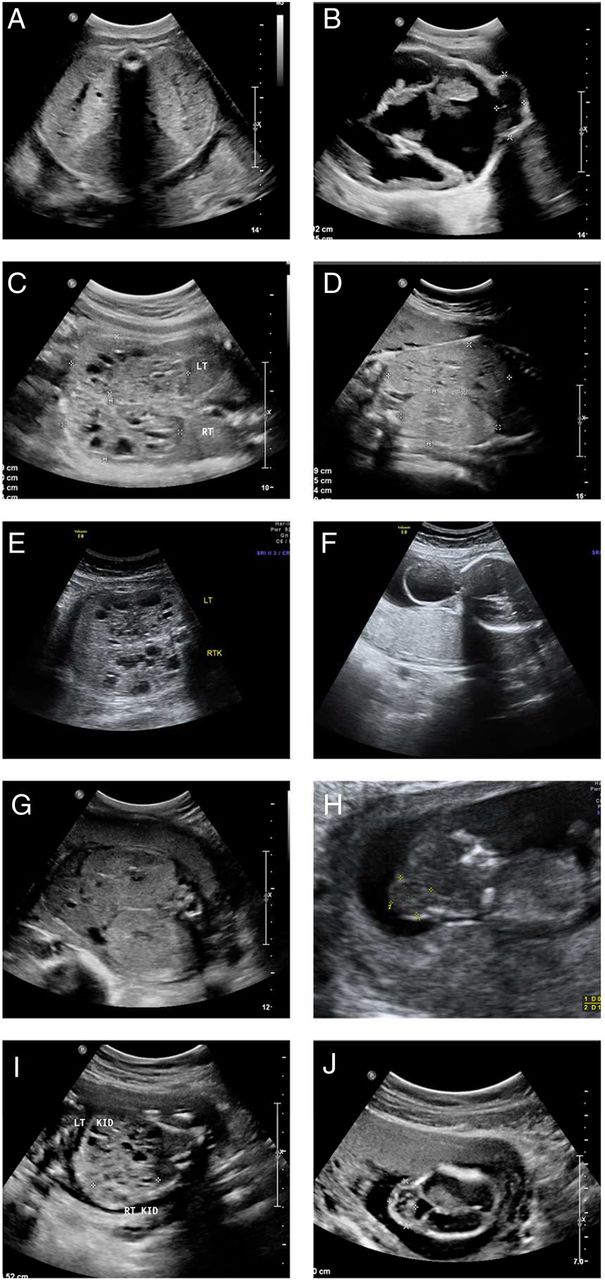
Prenatal ultrasound images of affected fetuses. (A and B) FT-8: transverse view of the fetal abdomen at 33 weeks of gestation (A), showing enlarged echogenic kidneys. Transverse view of the fetal head at 33 weeks of gestation (B), showing a cystic mass arising from the occipital area of the fetal head representing an encephalocele. (Genotype: CC2D2A homozygous mutation.) (C) FT-10: a transverse view of the fetal abdomen at 23 weeks of gestation, showing enlarged echogenic kidneys with cystic areas. (Genotype: TMEM67 homozygous mutation.) (D) FT-20: a transverse view of the fetal kidneys at 33 weeks, showing enlarged echogenic kidneys. (Genotype: TCTN2 homozygous mutation.) (E and F) FT-1: a transverse view of the fetal kidneys at 31 weeks of gestation (E), showing enlarged echogenic cystic kidneys. A transverse view of the fetal head at 31 weeks of gestation (F), showing a cystic mass arising from the fetal occiput, which represents an encephalocele. (Genotype: CC2D2A homozygous mutation.) (G) FT-9: a transverse view of the fetal abdomen at 27 weeks of gestation showing bilateral enlarged echogenic kidneys. (Genotype: INVS homozygous mutation.) (H) FT-13: a sagittal view of a fetus at 12 weeks of gestation showing a mass arising from the posterior aspect of the fetal head, which represents an encephalocele. (Genotype: MKS1 homozygous mutation). (I) FT-22: a transverse view of the fetal abdomen at 18 weeks, showing enlarged kidneys with cystic changes. (Genotype: TMEM67 homozygous mutation.) (J) FT-21: a transverse view of the fetal head at 16 weeks of gestation, showing a mass arising from the posterior aspect of the fetal head, which represents an encephalocele. (Genotype: CC2D2A homozygous allele.)
Where patient DNA was available (group A, n=34), none of the cases had evidence of chromosomal aneuploidy (data not shown) and therefore malformation syndromes associated with renal cysts, such as trisomy 13 (Patau), trisomy 18 (Edward) and trisomy 21 (Down), were excluded.
Using the renal gene panel in this cohort, 96.98% coverage of target genes was achieved, with an average base coverage of >500. Twenty-one patients from 34 families in cohort A had a molecular genetic diagnosis (62% mutation detection rate). Seven families from the 10 families in cohort B had an inferred molecular genetic diagnosis by the finding of identical pathogenic alleles in both parents who were known to be consanguineous. The chances of the affected child inheriting both these alleles would be 25%. The identified rare alleles identified in these parental samples are listed in online supplementary table S2. In each of the families, there is only a single rare heterozygous change that was identified and confirmed using Sanger sequencing in both parents allowing a genetic diagnosis to be inferred (see online supplementary figure S1). All mutations identified were confirmed and segregation analysis was performed (including screening unaffected siblings) using Sanger sequencing. Mutations in genes B9D1, CC2D2A, CEP290, INVS, MKS1, NEK8, PKHD1, RPGRIP1L, TCTN2, TMEM67 and TMEM231 were identified (table 1) with a total of 13 novel variants detected in this study (table 2 and figure 2). Pathogenic rare (<1% MAF) sequence variants were not detected in the other renal panel genes, in particular digenic or oligogenic changes in renal ciliopathy genes were not seen. A common RPGRIP1L missense variant in its heterozygous state was identified as a third allele in two cases and is discussed below. Consistent with the lethal phenotypes seen in this cohort, mutations in B9D1, CC2D2A, CEP290, MKS1, RPGRIP1L, TCTN2, TMEM67 and TMEM231 are all known to cause a MKS phenotype. Mutations in INVS and NEK8 have been reported in severe neonatal forms of NPHP with numerous extrarenal features.35 Phenotypes in some of the patients with mutations with PKHD1 mutations were comparatively less severe, accounting for three of the cases which survived beyond the perinatal period.
In silico analysis of novel mutations

{kind=link}
{kind=link}
Novel mutations identified in cohort with antenatal cystic kidney disease and ciliopathy phenotypes. (A) Three nonsense (B), four splice site (C), three frameshift (D), two missense and (E) one deletion novel mutations (boxed) were detected homozygously in patients or heterozygously in both parents with ciliopathy phenotypes. (Just one parental chromatogram is shown but a comparison of maternal and paternal chromatograms is shown in online supplementary figure S1). Family number (FT) is shown as well as mutation and predicted translational changes. Healthy control sequence is shown alongside. Intron–exon boundaries are marked with a vertical dashed line.
In group A (34 families), homozygous mutations were detected in 20 families with just one family with compound heterozygous mutations (FT-19), in keeping with the known high rates of consanguinity. In group B (10 families), homozygous mutations were inferred by finding identical heterozygous variants in both parents in seven cases, consistent with the known parental consanguinity. These mutations were presumed to be found in their homozygous state in the affected patient. Unfortunately, direct sequencing of patient DNA or any unaffected siblings was not available in these cases. All mutations detected were either previously reported (and known to be pathogenic) or novel and predicted to be pathogenic by using in silico scores (table 2). Novel mutations were all homozygous (or inferred to be homozygous from parental samples) and included predicted missense, frameshift, nonsense and splicing defects. The types of mutations detected in this cohort seem to correlate closely with the phenotypes observed. Most mutations detected were truncating, frameshift and splice site mutations. These mutations were often lethal, causing fetal death or perinatal death. In this study, six missense mutations resulted in fetal or perinatal death.
Mutations in CC2D2A gene were the most common cause of antenatally detected cystic kidney disease in our cohort, accounting for eight cases. All patients with CC2D2A mutations had severe CNS abnormalities; six had evidence of an encephalocele indicative of a MKS phenotype and two had evidence of CVA with a dilated cisterna magna, in keeping with a JBTS phenotype. Typical MKS phenotypes were seen in three patients with MKS1 mutations, which included two novel changes (table 2). The novel nonsense MKS1 mutation (p.Q356*) was present in two families (FT-5 and FT-13), suggesting that these families were related. In family FT-18, the TMEM231 mutation (p.V251I) led to perinatal death in two fetuses. The mutation is predicted to cause a splicing defect (table 2), due to its position as the last nucleotide in exon 4 of the TMEM231, although it appears to be a missense change.
PKHD1 gene mutations were found in four families. One case (FT-31) had associated lung hypoplasia and cardiac malformations and another (FT-33) had evidence of intrahepatic cysts (table 1). It has been reported that truncating mutations in PKHD1 gene may be lethal.12 In this study, mutations detected in PKHD1 gene were homozygous missense mutations rather than truncating mutations, one of which was novel (c.3539G>A; p.G1180E) and led to a perinatal death in the proband and a sibling (FT-45). Three families shared the c.4870C>T (p.R1624W) mutation, which has been reported previously (also in its homozygous state) in a Saudi Arabian patient, with a ‘later-onset’ ARPKD phenotype.30
Despite known consanguinity, compound heterozygous variants in RPGRIP1L were identified in family FT-19, with four affected siblings. The first variant c.640G>A (V214I) (rs139067427) is rare with a reported allele frequency of 0.05% and is predicted to be pathogenic (table 2). The second RPGRIP1L variant c.685G>A (p.A229T) (rs61747071) was identified in this family following Sanger sequencing of exon 6 of RPGRIP1L, as MAF filtering via our data pipeline had excluded this relatively common variant (MAF of 3.7%). The pathogenicity of this variant has been previously explored,34 and the variant has been shown to compromise the interaction of RPGRIP1L with RPGR. To determine the frequency of this allele in our patient cohort, Sanger sequencing of RPGRIP1L exon 6 was performed. The rs61747071 variant was also present heterozygously in affected patients from FT-8 (with a homozygous CC2D2A missense mutation) and FT-10 (with a homozygous TMEM67 missense mutation). The additional pathogenicity of this allele in these patients is unknown.
Compound heterozygous mutations in B9D1 have previously been associated with MKS,36 but this gene remains a rare cause of renal ciliopathies. The fetus in this case presented with posterior encephalocele and bilaterally enlarged multicystic dysplastic kidneys and bilateral clubfeet (but not polydactyly). An additional disease allele in CEP290 identified in the fetus may have modified the phenotype.36 The B9D1 protein has structural similarities to MKS1 and similar severe phenotypes would be predicted. More recently, mutations in B9D1 have been described in two unrelated patients (aged 7 and 9 years) and with JBTS and a neurological limited phenotype, suggesting a wider phenotypic spectrum.37
Mutations in NEK8 are also a rare cause of a renal ciliopathy. Previously, homozygous mutations in NEK8 have been described in a Kurdish child with kidney microcysts and likely NPHP, reaching ESRD at 14 years of age (c.1273C>T, p.H425Y)38 and in three stillborn fetuses with enlarged cystic kidneys and cystic changes in the liver and pancreas (c.1795C>T, p.R599*).35 Some fetuses had additional features including heterotaxy, truncus arteriosus and other structural heart defects, hypoplastic lungs and skeletal anomalies (bowed femurs). Here, we identified a single stillborn fetus (FT-36) with a novel nonsense change in NEK8 (c.1401G>A, p.W467*) who had cystic kidneys, oligohydramnios, CVA and bilateral bowing of the femurs. This nonsense mutation is predicted to disrupt the highly conserved regulator of chromatin condensation 1 (RCC1) domain and is in proximity to the murine jck mutation (p.G448V).39
Antenatal presentations of cystic kidney disease are often associated with severe phenotypes and poor outcomes. These can include early presentation of ADPKD, or more commonly in consanguineous families, a presentation of an autosomal-recessive renal ciliopathy disease, as we have seen in this cohort. Extrarenal manifestations on the antenatal USS such as encephalocele and CVA may suggest MKS or JBTS phenotypes, respectively. Other features such as polydactlyly and thoracic cage abnormalities may point towards other ciliopathies such as BBS40 or skeletal dysplasias such as Jeune syndrome.41
Screening for ciliopathy genes in the diagnostic setting, especially in the perinatal period, is challenging. While whole exome sequencing (WES) is one possible approach, targeted gene panel exome sequencing may be preferable in diagnostic laboratories for specificity, deliverability and low cost. A disease-specific gene panel approach avoids the common difficulty of reporting secondary genetic findings that often occurs following WES. However, any predesigned NGS gene panel will be limited to known genes directed towards specific phenotypes and will not allow for recently discovered ciliopathy genes to be screened. Our gene panel contained 90 genes and included the 3 known polycystic kidney disease genes (PKD1, PKD2 and PKHD1) and 11 of the 12 known MKS genes (see online supplementary table S1). However, it included only 10 of the 21 genes known to cause NPHP and 9 of the 26 JBTS genes. Therefore, our panel was biased towards (and very effective at) diagnosing MKS in this cohort with very severe disease phenotypes, but the precise molecular genetic diagnosis remained unknown in others. Indeed, in the 14 cases whom had antenatal USS evidence of an encephalocele suggestive of a MKS phenotype, all except 2 had a molecular genetic diagnosis. A recent study confirmed the strong cystic kidney disease phenotype in MKS patients, where cystic kidneys were found in (97.7%) of MKS cases.22
To improve diagnostic yield, unsolved samples via our panel gene testing could be subjected to WES, especially in cases where there are DNA samples available from more than one affected in each family. However, this approach is more costly and can be more time consuming, when compared with a targeted panel approach. We hope to develop an updated renal gene panel in the near future, as the NGS sequencing platform we have developed will allow for additional genes (and their amplicons) to be analysed. In this study cohort of antenatal cases, the mutation detection rate was higher than reported by others36 ,42 who used PCR exon sequencing alone. More recently, a combination of WES and targeted resequencing of a ciliopathy gene panel was successfully used in a cohort of patients with Jeune asphyxiating thoracic dystrophy.43
In summary, using a cohort of patients with antenatal evidence of kidney disease and associated ciliopathy syndromes, we have performed targeted genetic panel testing using patient and/or parental DNA samples to reveal the molecular genetic diagnosis in 64% of patients. Our high detection rate of homozygous disease-causing alleles reflects a high underlying rate of consanguinity. We would predict a reduction in diagnostic yield in less consanguineous populations. The genetic spectrum remains wide and certainly we have not identified a reason to narrow our diagnostic panel, rather it should be expanded to capture more recently reported genetic causes of developmental renal disease. It is interesting to note that CC2D2A mutations were the commonest cause of an antenatal ciliopathy in our cohort, but the genetic heterogeneity of inherited cystic kidney disease is also borne out by our study. In our population, renal gene panel testing provided diagnostic information that was valuable to clinicians, genetic counsellors and families. A molecular genetic diagnosis provides an accurate diagnosis, which is hugely valuable when there are such severe phenotypes affecting one or more family members and can be used to predict recurrence rates and allow planning, including preimplantation genetic diagnosis for future pregnancies.
Acknowledgments
We thank all the families for participating in the study. We also thank all team members at Saudi Human Genome project for help in conducting the study. In addition, we thank sequencing and genotyping core facilities at Genetics Department at the Research Centre, KFSH&RC for doing sequencing and molecular karyotyping. Special thanks for Ms Ghadah A Al Dakheel for preparing figures for publication. JAS acknowledges support from the Northern Counties Kidney Research Fund. All NGS library building, sequencing and bioinformatics analyses were performed by the Saudi Human Genome Project at King Abdulaziz City for Science and Technology (KACST) and at KFSHRC.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors MHA-H, JAS and BM conceived of the study and participated in its design and coordination, and drafted and revised the manuscript. BAA, FSB, DK, DM and NA performed the NGS sequencing. AA and NE performed in silico modelling. HA-J, AA, AIT, DK, NE, BAA and FSB carried out all technical aspects of molecular diagnosis and helped with in silico modelling. WK, NA-S, ZA, RA, MT, MA and RK participated in the clinical diagnosis of the cases. SM analysed molecular karyotyping. TF, ME-K and MA carried out bioinformatics analysis. DM and NA-T helped conceive the study and drafted the manuscript. All authors read and approved the final manuscript.
Competing interests None declared.
Ethics approval Research Advisory Council of King Faisal Specialist Hospital, Riyadh, Saudi Arabia (RAC#2050 045).
Provenance and peer review Not commissioned; externally peer reviewed.